Phylogenomic relationships and molecular convergences to subterranean life in rodent family Spalacidae
2021-10-18Yuan-TingGuo,JiaZhang,Dong-MingXu等
All extant species in the rodent family Spalacidae are subterranean and have evolved various traits for underground life. However, the phylogenomic relationships among its three subfamilies (Myospalacinae, Spalacinae, and Rhizomyinae)and the molecular basis underlying their adaptations to underground life remain poorly understood. Here, we inferred the phylogenomic relationships among these subfamilies based onde novosequencing the genome of the hoary bamboo rat (Rhizomys pruinosus). Analyses showed that~50% of the identified 11 028 one-to-one orthologous proteincoding genes and the concatenated sequences of these orthologous genes strongly supported a sister relationship between Myospalacinae and Rhizomyinae. The three subfamilies diversified from each other within ~2 million years.Compared with the non-subterranean controls with similar divergence dates, the spalacids shared more convergent genes with the African subterranean mole-rats at the genomic scale due to more rapid protein sequence evolution.Furthermore, these convergent genes were enriched in the functional categories of carboxylic acid transport, vascular morphogenesis, and response to oxidative stress, which are closely associated with adaptations to the hypoxichypercapnic underground environment. Our study presents a well-supported phylogenomic relationship among the three subfamilies of Spalacidae and offers new insights into the molecular adaptations of spalacids living underground.
All living species in the rodent family Spalacidae are exclusively subterranean and have evolved a variety of similar traits to other subterranean mammals to adapt to their underground conditions (Emerling & Springer, 2014; Nevo,1979). Given their phenotypic similarities, morphological characters alone limit the unambiguous reconstruction of phylogenetic relationships among the three subfamilies within Spalacidae, i.e., Myospalacinae (zokors), Spalacinae (blind mole-rats), and Rhizomyinae (bamboo rats) (Lin et al., 2014;Zou & Zhang, 2016). Different molecular markers, as well as transcriptomic data, have been used to establish phylogenetic trees of these subfamilies (He et al., 2020; Jansa & Weksler,2004; Jansa et al., 2009; Lin et al., 2014). However,inconsistent tree topologies suggest that the phylogenetic relationships among zokors, blind mole-rats, and bamboo rats should be investigated at the genomic scale. In addition, the spalacids and African mole-rats (e.g., naked mole-rats and Damaraland mole-rats) in rodents, as well as several other mammalian lineages, have evolved independently for underground life, with a variety of convergent phenotypic changes (Nevo, 1979, 1995). Examining convergent evolution at the molecular level is a useful means to investigate the molecular basis of convergent phenotypes (Stern, 2013; Storz,2016), including subterranean mammals (Davies et al., 2018).At present, systematic examination of the molecular convergences underlying the complex convergent phenotypes of underground adaptations on the ancestral and descendant branches of spalacids is limited at the genomic scale. Such studies may provide insights into the molecular basis of phenotypic adaptations to the subterranean niche in mammals.
Among the three Spalacidae subfamilies, genomic data are available for the plateau zokor (Eospalax baileyi) in Myospalacinae and the blind mole-rat (Nannospalax galili) in Spalacinae. However, no genomic data are currently available for any species of the subfamily Rhizomyinae. To reconstruct the phylogenomic relationships among these subfamilies, we selected the hoary bamboo rat (Rhizomys pruinosus) as a representative species of Rhizomyinae and sequenced its genome using the Illumina Hiseq 2500 and PacBio platforms.After obtaining a total of 728.62 Gb and 33.17 Gb of clean data (>205×) from the two platforms, we yielded a draft assembly with contig and scaffold N50 sizes of 46.61 kb and 2.2 Mb, respectively. A total of 3 476 out of 4 104 (84.7%)conserved mammalian genes were assembled completely(Supplementary Tables S1-S4). Overall, genome quality was comparable to that of other subterranean and nonsubterranean rodents (Supplementary Table S5) and was sufficient to infer phylogeny and perform analysis at the genomic scale.
Combined with published genomic data, we identified a total of 11 028 one-to-one orthologous protein-coding genes across the hoary bamboo rat, plateau zokor, blind mole-rat, and mouse (Mus musculus), which was used as the outgroup of the spalacids. Before phylogenetic analysis, we evaluated the potential effects of sequence saturation by performing the substitution saturation test and found that the orthologous protein-coding genes experienced little substitution saturation(two-tailedP<0.001). For each orthologous protein-coding gene, both Bayesian inference (BI) and maximum-likelihood(ML) approaches were used to reconstruct the phylogenetic relationships among the hoary bamboo rat, plateau zokor,blind mole-rat, and mouse based on the best-fitting substitution model estimated by jModelTest2 (Darriba et al.,2012). Among the phylogenetic trees with >0.9 Bayesian posterior probability (BPP) and >90 bootstrap support (BSS),about half (45.1% and 51.5%) supported a sister group relationship between the hoary bamboo rat and plateau zokor(termed H1), ~20% supported a sister group relationship between the hoary bamboo rat and blind mole-rat (H2), and~30% supported a sister group relationship between the plateau zokor and blind mole-rat (H3) (Figure 1A;Supplementary Table S6). These results were robust when examining tree topologies with different supporting cutoffs of>0.8, >0.7, >0.6, and >0.5 BPP and >80, >70, >60, and >50 BSS (Figure 1A).
We also reconstructed the phylogenomic tree for hoary bamboo rat, plateau zokor, blind mole-rat, and mouse by concatenating all 11 028 one-to-one orthologous proteincoding genes. Using partitioned ML and BI approaches,phylogenetic analysis of the concatenated orthologous sequences strongly supported a sister group relationship between the bamboo rat and plateau zokor with high supporting values at all nodes (1.0 BPP and 100 BSS), in accordance with the above phylogenetic topology of individual genes. Based on the reconstructed phylogeny above, we estimated divergence times among Rhizomyinae,Myospalacinae, and Spalacinae. A total of 2 491 914 four-fold degenerate sites were extracted from the 11 028 one-to-one orthologous protein-coding genes. Based on these sites, we used the Monte Carlo Markov Chain algorithm (Yang, 2007)with three fossil calibration points and estimated that the spalacids shared a common ancestor with mice ~46.1 million years ago (Mya) (Figure 1B). The blind mole-rats first diverged from the common ancestor of spalacids ~27.9 Mya; ~2.1 million years later the bamboo rats and zokors diverged(Figure 1B).
To reveal the molecular basis of subterranean life in spalacids, we investigated genome-wide convergent evolution between spalacids and other subterranean rodents. In total,6 674 one-to-one orthologous protein-coding genes were obtained across 12 mammals, including two African mole-rats,i.e., naked mole-rat (Heterocephalus glaber) and Damaraland mole-rat (Fukomys damarensis), which are phylogenetically distant subterranean rodents of the spalacids (Figure 1C). We first examined the genome-wide protein-sequence convergences between the last common ancestor (LCA) of the African mole-rats (Branch I) and the LCA of the spalacids(Branch III). A total of 95 convergent amino acids in 95 genes and 146 divergent amino acids in 143 genes were identified between branches I and III (Supplementary Tables S7, S8).As a comparison, we searched for convergent and divergent amino acids between the LCA of the non-subterranean degus and guinea pig (Branch II) and branch III (Supplementary Tables S9, S10). Due to nearly identical divergence dates between the degus and guinea pig, and between the two African mole-rats (Jiang et al., 2020), the LCA of the degus and guinea pig should be suitable as a non-subterranean control branch for the LCA of the African mole-rats(Figure 1C). The number of convergent sites is considered to be proportional to the number of divergent sites under no adaptive convergence (Castoe et al., 2009); thus, adaptive convergent evolution was tested by examining whether the number of convergent sites between branches I and III significantly exceeded that between branches II and III when controlling their respective divergent site numbers. As a result,the ratio of the number of convergent sites to the number of divergent sites (95/146) between branches I and III was not significantly larger than that (70/107) between branches II and III (P=0.98; two-tailedχ2test). Next, we examined genomewide protein sequence convergences between branch I and each of the three subfamilies of spalacids, as described above. If a gene contained convergent substitutions on the ancestral branch pairs (branches I and III or II and III), the molecular convergences were not counted again in the tip comparisons. The convergent to divergent site ratios from the three branch pairs of subterranean rodents (i.e., branches I vs.IV, I vs. V, and I vs. VI) were not significantly different from those of their respective control branch pairs (i.e., branches II vs. IV, II vs. V, and II vs. VI) (Supplementary Tables S11-S22).
Although there were no significant differences in the convergent to divergent site ratios, there were generally more convergent genes between spalacids and subterranean African mole-rats than between their control pairs at the genomic scale (P<0.05; two-tailedχ2tests; Figure 1D). To explore the molecular basis of the subterranean lifestyle in spalacids from these convergent genes, we performed Gene Ontology (GO) functional enrichment analysis of the 95 convergent genes between branches I and III using all one-toone orthologous protein-coding genes as the background. A total of 111 biological process (BP) terms were significantly enriched (uncorrectedP<0.05; Supplementary Table S23),including six (among the top 10) terms related to response to biotic stimulus, which is largely consistent with previous research (Davies et al., 2018). In comparison, GO analysis was performed for the 68 convergent genes between branches II and III, and no BP terms related to biotic stimulus were found among the significantly enriched functional categories (Supplementary Table S24).
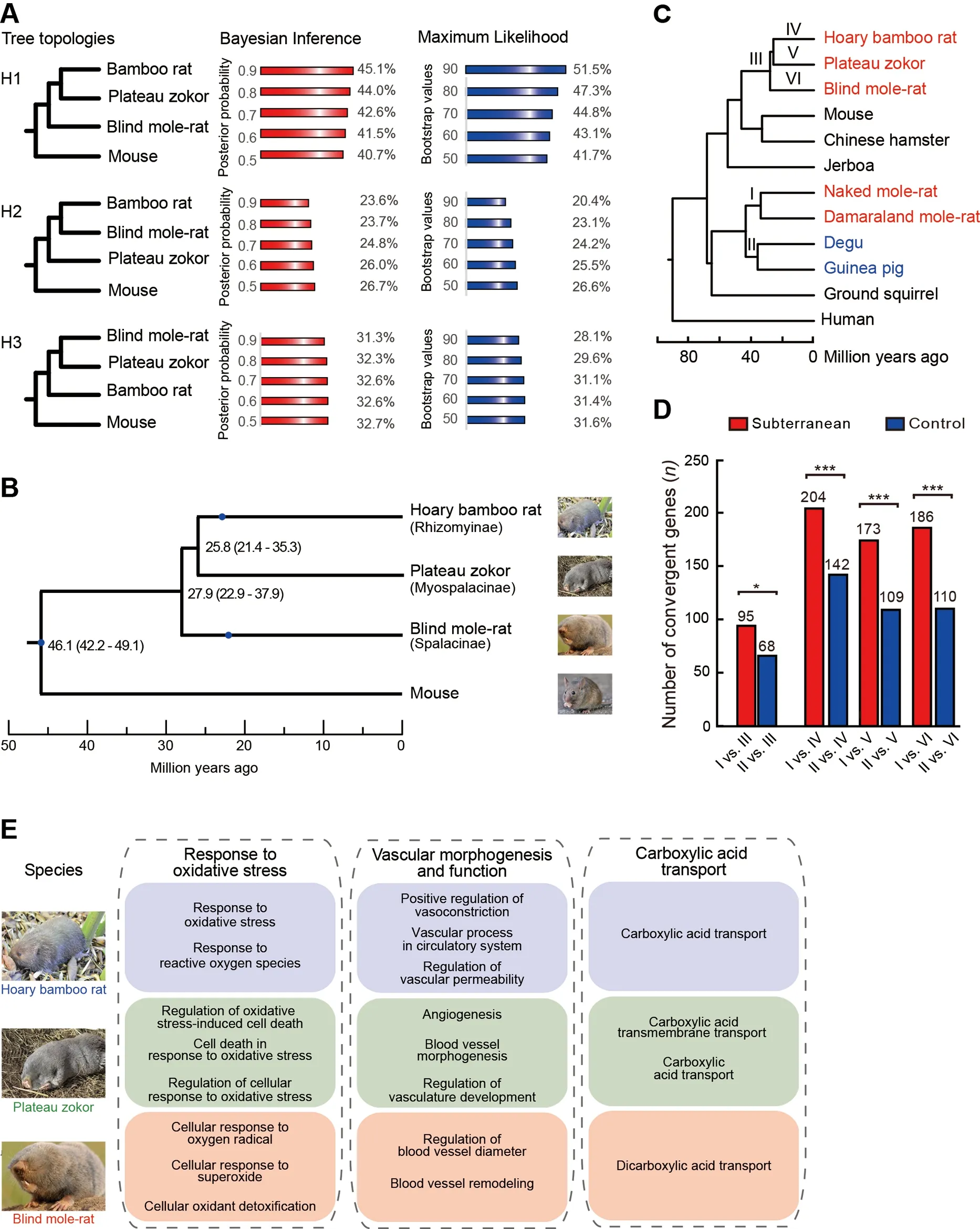
Figure 1 Reconstruction of phylogeny among spalacids and genome-wide convergent analyses
We next conducted functional enrichment analysis of the convergent genes between the African mole-rats and each of the three subfamilies of spalacids (i.e., branches I vs. IV, I vs.V, and I vs. VI). A total of 85, 119, and 56 significantly enriched BP terms were identified, respectively (uncorrectedP<0.05; Supplementary Tables S25-S27). Although there were few shared GO terms, several similar functional categories among the three datasets were found and may be related to life underground, especially to low oxygen and high carbon dioxide levels, including response to oxidative stress,vascular morphogenesis and function, and carboxylic acid transport (Figure 1E). These shared functional categories were not found for the same enrichment analyses of convergent genes among the control branch pairs(Supplementary Tables S28-S30). Notably, despite similar functional annotations, direct evidence is still needed to prove that these functional categories are the molecular basis of spalacids living underground.
DATA AVAILABILITY
RNA-seq data were deposited both in the NCBI under BioProject No. PRJNA573514 and in the Genome Sequence Archive (GSA) database under Accession No. CRA004915.The whole genome sequence data in this paper have been deposited in GenBank under accession No. VZQC01000000 and also deposited in GSA under accession No.GWHBEDE00000000.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Z.L. and L.Z.T. designed the study. L.Z.T. collected the sample and extracted the genomic DNA. Y.T.G., J.Z. and D.M.X. assembled and annotated the genome and analyzed the data. Y.T.G. and Z.L. wrote the draft manuscript. Y.T.G.and Z.L. revised the manuscript. All authors read and approved the final version of the manuscript.
杂志排行

Zoological Research的其它文章
- Prosecution records reveal pangolin trading networks in China, 2014-2019
- Chromosome-scale genome assembly of brownspotted flathead Platycephalus sp.1 provides insights into demersal adaptation in flathead fish
- Deletion of phosphatidylserine flippase β-subunit Tmem30a in satellite cells leads to delayed skeletal muscle regeneration
- Role of juvenile hormone receptor Methoprene-tolerant 1 in silkworm larval brain development and domestication
- Captopril alleviates lung inflammation in SARS-CoV-2-infected hypertensive mice
- Global view on virus infection in non-human primates and implications for public health and wildlifeconservation