复发型多发性硬化症疾病修饰治疗新药
——庞西莫德(ponesimod)
2021-10-09陈本川
陈本川
(湖北省医药工业研究院有限公司,武汉 430061)
多发性硬化症(multiple sclerosis,MS)是一种慢性中枢神经系统(central nervous system,CNS)自身免疫性炎症性疾病,多发于20~40岁青年人群,影响全球230万人,女性发病率高于男性。其特征是自身免疫T 细胞破坏血脑屏障,炎症递质进入并攻击CNS,通过炎症和组织损伤,破坏脱髓鞘和轴突丢失,导致大脑、视神经和脊髓的正常功能衰减和严重致残。MS的主要亚型为复发型多发性硬化症(remitting multiple sclerosis,RMS),占MS的85%,包括临床孤立综合征(clinically isolated syndrome,CIS)、复发-缓解型多发性硬化症(relapsing-remitting multiple sclerosis ,RRMS)、继发性进行性多发性硬化症(secondary progressive multiple sclerosis,SPMS)和原发进展型多发性硬化症(primary progressive multiple sclerosis,PPMS)。SPMS的定义为新的、恶化的或复发的神经症状,持续>24 h,无发热或感染。复发可能在数天或数周内完全缓解,或导致持续的残缺和残疾的累积。近期,鞘氨醇-1-磷酸(sphingosine-1-phosphate,S1P)受体抑制药已成为MS领域新药研发的一个重要靶点,但也无法治愈MS,只能减少复发症状的频率。第一个获得美国食品药品管理局(FDA)批准上市的S1P受体抑制药品种是由瑞士诺华制药公司研制盐酸芬戈莫得(figolimod hydrochloride)胶囊,于2010年9月21日批准上市,商品名为Gilenya®,用药1 年,复发率为18%,95%CI=(0.16,0.21)%。第2个品种是2012年9月12日FDA 批准上市法国Sanofi-aventis制药集团公司研制特立氟胺(teriflunomide),片剂的商品名Aubagio®,2018年国内有多家制药企业引进仿制,每天1次,口服特立氟胺7或14 mg,分别降低复发风险31.2%和31.5%,减少MS患者病情持续恶化的比例为21.7%和20.2%。此外,两个剂量组特立氟胺均能显著改善MS患者的脑部病变,与安慰药组比较,特立氟胺2个剂量组年复发率均为37%,特立氟胺降低MS患者年复发风险率分别为31.2%和31.5%;残疾进展比例分别为21.7%和20.2%。第3个品种是瑞士诺华公司研制西尼莫德(siponimod),FDA于2019 年3月26日批准上市,欧洲药物管理局(EMA)于2020年1月13日批准上市,片剂商品名为Mayzent®。在为期24个月试验,每天分别服西尼莫德2.0或10 mg,年复发率为0.20%,95%CI=(0.10,0.38)%和0.22%,95%CI=(0.12,0.40)%;在长达24个月治疗中,MS的病变活性低。MRI结果显示,2和10 mg剂量组均具有持续的疗效。由瑞士Actelion生物制药公司研制ponesimod,暂译名为庞西莫德,亦译为珀奈莫德、泊沙莫德和泊尼莫得等。英文化学名为(2Z,5Z)-5-[3-chloro-4-[(2R)-2,3-dihydroxypropoxy]benzylidene]-3-(2-methylphenyl)-2-(propylimino)-1,3-thiazolidin-4-one。中文化学名(2Z,5Z)- 5-[3-氯-4-[(2R)- 2,3-二羟基丙氧基]亚苄基-3-(2-甲基苯基)-2-(丙基亚氨基) )-1,3-噻唑烷-4-酮。代号为ACT128800。2017年1月,强生公司以300亿美元,将Actelion公司的庞西莫德及多个正在研究候选品种纳入强生公司的全资子公司,并责成其控股的杨森制药公司全力开发庞西莫德;2019年9月杨森公司在瑞士召开欧洲MS治疗与研究委员会第35届会议上公布新型口服选择性S1P1受体调节药庞西莫德的Ⅲ期临床研究结果。该研究在RMS成人患者中开展,将庞西莫德与法国赛诺菲制药公司治疗MS新药特立氟胺进行头对头的对比试验。庞西莫德在临床主要观察指标或次要指标均取得优异的疗效。2020年3月22日杨森公司向美国FDA递交新药上市申请(NDA)。2020年3月4日杨森公司向EMA提交申请营销授权应用(MAA),以寻求批准用于治疗成人RMS。2021年3月18日FDA批准庞西莫德片剂上市,商品名为Ponvory®,用于治疗成人RMS。经庞西莫德片治疗的成人RMS患者,年复发率比特立氟胺治疗组降低近1/3,庞西莫德片成为第一个,也是目前唯一获得FDA批准的针对现有口服S1P受体抑制药的疾病修饰疗法(disease modifying therapy,DMT)新药。EMA属下的人用医药产品委员会(CHMP)也发布一份积极审查意见,建议EMA尽快批准庞西莫德片。2021年5 月 24 日批准在欧盟所有27 个成员国和3个欧洲经济区国家:挪威、冰岛和列支敦士登可临床使用庞西莫德治疗成人RMS[1-4]。
1 非临床药理毒理学
1.1致畸、致突变 分别给雄性小鼠喂饲庞西莫德0,50,150或400 mg·kg-1·d-1或给雌性小鼠喂饲庞西莫德30,100或300 mg·kg-1·d-1,为期2年,雄、雌小鼠在所有含药的剂量组均产生血管肉瘤,最高剂量组雄、雌小鼠都产生血管瘤及血管肉瘤。给雄小鼠喂饲最低剂量庞西莫德50 mg·kg-1·d-1时,血浆药物接触量,即血浆药物浓度-时间曲线下面积(AUC)约为人用推荐剂量(recommended dose for human use,RHD)20 mg的5倍。分别给雄大鼠喂饲庞西莫德0,3,10或30 mg·kg-1·d-1,或给雌性大鼠喂饲庞西莫德0,10,30或100 mg·kg-1·d-1,为期2年,不会导致肿瘤的增加。雄大鼠接触庞西莫德最大剂量30 mg·kg-1·d-1,相当于RHD 20 mg的4倍。庞西莫德无基因毒性,体外Ames试验、哺乳动物染色体畸变试验和和体内大鼠微核试验均为阴性[1,3-4]。
1.2对生殖能力的影响 在隔离研究中,大鼠交配前、整个交配期,直至雌大鼠妊娠第6天,分别给雄、雌大鼠喂饲庞西莫德0,10,30或100 mg·kg-1·d-1,对大鼠生育能力无影响。血浆中庞西莫德的接触量,按AUC估算,雄大鼠最高剂量组约为RHD 20 mg·d-1的20倍;雌大鼠约为RHD的30倍[1,3-4]。
1.3动物毒理学和(或)药理学实验 对小鼠、大鼠和犬进行庞西莫德口服毒性实验研究中,观察到动物肺体质量和组织病理学改变(包括肺泡组织细胞增生、水肿)。短期给予大剂量庞西莫德,观察到肺泡组织细胞增多、肺水肿、肺气肿或透明质病。停药后,大鼠出现支气管肺泡增生和犬肺泡组织细胞增多及透明质病。长期服药后,疗效往往不明显或病变较轻。这些发现认为是S1P1受体引起的血管通透性增加次要因素的调制作用。未观察到损害作用的剂量浓度(no observed adverse effect level,NOAEL)。在对大鼠和犬为期4周口服庞西莫德毒性研究中,肺功能的影响与AUC相似,或低于预期的RHD[1,3-4]。
2 临床药理毒理学
2.1作用机制 庞西莫德是一种S1P1受体的调节药,在淋巴细胞表面上表达,对S1P1受体具有高亲和力,能阻断淋巴细胞从淋巴结排出,减少外周血中淋巴细胞的数量。庞西莫德发挥治疗MS的作用机制尚不清楚,可能涉及减少淋巴细胞迁移到中枢神经系统有关[1,3-4]。
2.2药效学
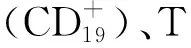
2.2.2对心率与心律的影响 庞西莫德引起心率和房室传导短暂降低,并呈剂量正相关性,应延迟开始治疗。心率下降在庞西莫德剂量≥40 mg时达到稳定水平(为推荐服2倍剂量,以维护心率的稳定)。与安慰药比较,在以下情况下,可能检测到心律失常事件,如房室传导阻滞有较高的发生率。庞西莫德这种作用在给药的第1个小时内开始,给药后2~4 h达到最大值。心率一般会在用药后4~5 h恢复到用药前水平,第1天,重复给药,心率下降减弱,表明有耐受性。阿托品可以逆转由庞西莫德引起的心率下降。
2.2.3与β受体阻断药联用的影响 在专项药效学安全性研究中,庞西莫德与β受体阻断药普萘洛尔联用有明显的负变时效应,普萘洛尔达到稳态时,加服庞西莫德,对心率有叠加效应。
2.2.4心脏电生理学 在一项全程QT研究,每天分别服庞西莫德40和100 mg(推荐维持剂量的2倍和5倍),直到稳态,经fridericia校正的QT间期(QTcF)延长,90%CI双侧上限,最大均值40 mg 剂量组为11.8 ms,100 mg剂量组为16.2 ms。无1例受试者庞西莫德QTcF绝对值>480 ms或QTcF>90 ms。
2.2.5肺功能 经庞西莫德治疗的受试者,最大深吸气后,第一秒呼出气量的容积(FEV1)和用力肺活量(forced vital capacity,FVC)均比安慰药组大,且与剂量呈正相关。这些影响在服短效β2激动药后可逆转。
2.3药动学 口服庞西莫德1.0~75 mg,其血药峰浓度(Cmax)和AUC增加与剂量接近正相关。血浆稳态浓度约比单次服药浓度增加2.0~2.6倍。口服庞西莫德固定的维持剂量,3 d后可得到稳态。健康受试者口服庞西莫德的药动学相似,MS患者在不同学科的研究有25%的差异[1,3-4]。
2.3.1吸收 达到Cmax的时间(tmax)为服药后2~4 h。庞西莫德10 mg剂量绝对口服生物利用度为84%。食物对庞西莫德的药动学无临床相关意义的影响,是否在进食时服药均可。
2.3.2分布 健康受试者静脉给药后,庞西莫德的稳态分布容积为160 L。庞西莫德与血浆蛋白有高度结合(>99%),全血的血浆主要分布于血浆中(78.5%)。动物研究表明,庞西莫德很容易穿越血-脑屏障。
2.3.3代谢 庞西莫德达到推荐剂量的浓度,在人体排泄前已被广泛代谢,尽管成分没改变,血浆中主要循环成分已在人血浆中鉴定出两种非活性循环代谢物,M13和M12,分别约占药物相关接触总量的20%和6%。两种代谢产物均对S1P受体无活性。人肝制剂的实验表明,庞西莫德可以代谢为M13主要通过非细胞色素P450(CYP)酶的组合发生代谢活动。多个CYP(CYP2J2,CYP3A4,CYP3A5,CYP4F3A和CYP4F12)和非CYP酶催化将庞西莫德氧化为M12。庞西莫德也直接受到葡萄糖醛酸化,主要是UGT1A1和UGT2B7。
2.3.4排泄 单次静脉注射庞西莫德后,总清除率为3.8 L·h-1。口服给药后,消除半衰期(t1/2)约33 h。单次口服14C-庞西莫德后,从大便回收放射性剂量57%~80%,其中16%为原形药;从尿液回收10%~18%放射性剂量,无原形药。
2.3.5特殊人群的药动学 ①年龄在17~65岁,对庞西莫德的药动学参数无明显影响,≥65岁老年患者对庞西莫德药动学参数的影响尚不清楚。性别和种族对庞西莫德药动学参数的影响无临床意义。②肾损伤患者无需调整剂量:成年中度肾损伤患者肌酐清除率(CrCl)Cockroft-Gault估计值CrCl=30~59 mL·min-1和重度肾损伤患者CrCl<30 mL·min-1,与正常肾功能患者比较,庞西莫德Cmax和AUC无明显变化。正在透析的患者对庞西莫德药动学的影响尚不清楚。因庞西莫德血浆蛋白结合率>99%,不需要透析,预计会改变庞西莫德总浓度和未结合的浓度,故不需要调整剂量。③肝损伤患者为轻度、中度或严重肝损伤成人患者(Child-Pugh分别为A级、B级和C级)对庞西莫德的Cmax无影响,但庞西莫德的AUC0 -∞与健康受试者比较,分别增加1.3,2.0和3.1倍。
3 临床试验
3.1临床试验概况 开发公司计划对庞西莫德片开展14批临床试验,用于验证治疗MS、牛皮癣和抗移植物抗排异等适应证的疗效,纳入3584例受试者,其中,Ⅰ期临床6批227例;Ⅱ期临床5批1211例;Ⅲ期临床试验3批2146例。用于验证治疗MS的临床试验,有11批次,纳入3191例受试者,其中Ⅰ期临床6批227例;Ⅱ期临床2批818例;Ⅲ期临床试验3批2146例。在美国FDA批准庞西莫德片上市之际,所有临床试验接近全部完成,少数批次试验正处于数据处理阶段,开发公司也在期刊上公开发表一份Ⅲ期临床试验结果[1,4-5]。
3.1.1临床实验入选标准 ①开始任何研究授权程序前,必须签署知情同意书。②年龄在18~55岁男、女受试者,确诊符合MS诊断标准2010修订版,受试者有复发性发作,即RRMS和SPMS,必须患有活动性疾病,其通过随机分组前12个月内 MS发作≥1次,或在随机分组前24个月内有≥2次MS发作的证据。③在随机分组前6个月内,通过磁共振成像(magnetic resonance imaging,MRI)对大脑钆离子(Gd+)增强病变进行检查。④入选受试者必须是非卧床的。残疾状况评分量表(expanded disability status scale,EDSS)评分≥5.5,并且未经治疗或曾接受过MS疾病改良疗法治疗。⑤有生育能力的妇女接受治疗前尿液检测妊娠试验必须是阴性,并同意进行为期4周的尿液妊娠试验,且必须一直使用可靠的避孕方法。参加研究的有生育男性功能的受试者必须同意使用安全套。
3.1.2临床实验排除标准 (1)首次服药前有以下任何心血管疾病:①静息心率(HR)<50次·min-1;②存在二级或三级房室传导阻滞或QTcF间隔女患者>470 ms,男患者>450 ms。(2)试验核心研究(experimental core research,ECR)第14次随访时,中央实验室提出以下任何警报,已重复测试中确认为警报,或在试验核心研究完成前未重复进行:①淋巴细胞计数<0.2×109·L-1;②中性粒细胞计数<1.0×109·L-1;③血小板计数<50×109·L-1;④肌酐清除率<30 mL·min-1。(3)在核心研究第14次随访时,与研究基线比较,FEV1和(或)FVC降低>30%。(4)具有临床意义的持续性呼吸道不良事件(如呼吸困难),在扩展研究中首次给药前未缓解。(5)在核心研究首日的筛查与延伸研究的首日之间的任何时间出现黄斑水肿。(6)在核心研究的第14次随访或第108周试验研究完成或扩展研究的首日,存在以下情况:研究者认为,①CNS的可疑机会性感染或任何其他感染,有碍于药物重新开始研究的行为;②有史蒂文斯-约翰逊综合征(Stevens-Johnson syndrome)或中毒性表皮坏死溶解或嗜酸粒细胞增多症和全身症状的药物过敏性反应。(7)妊娠妇女或哺乳期妇女及治疗期间患者男性伴侣渴望生育孩子。(8)曾服用过任何MS疾病改良疗法进行治疗。(9)研究者认为,有任何其他与临床相关的医学或外科疾病会使受试者处于危险状态或受试者不太可能遵守扩展研究方案。
3.1.3临床疗效主要观察指标 年度确诊复发率(annual recurrence rate,ARR),时限为从核心研究(入组)的第1天到扩展研究的治疗结束,即长达354周。ARR的定义为每科目年确认的复发次数。复发的定义为新的、恶化的或复发的神经系统症状,在先前复发发生后至少 30 d出现,持续至少24 h,在无发烧或感染的情况下发生,并伴有记录的增加EDSS评分或其功能系统评分来自先前的临床评估。从基线起,扩展EDSS得分或1~3个功能系统至少增加0.5分,不包括肠、膀胱、脑、精神和小腿。EDSS和FS评分基于神经系统检查,评估其在MS中的损害。在8个系统中,有7个是序数临床评分量表,范围从0~5或6,以及较高的量表指示总体功能受损,评估视觉、脑干、锥体、小脑、感觉、肠/膀胱和脑功能。通过对各个系统评分进行汇总,结合有关步态和辅助使用的观察结果和信息,对EDSS进行评分。EDSS是序贯临床评分量表,范围0~10(MS死亡)
3.1.4临床疗效次要观察指标 ①通过疲劳症状和影响问卷调查的症状域-复发性多发性硬化症(fatigue symptoms and impact questionnaire -relapsing multiple sclerosis,FSIQ-RMS)的症状域来衡量,与疲劳相关的症状从基线到108周的变化,时限为从基线到第108周,治疗结束(end-of-treatment,EOT)。FSIQ-RMS是Actelion开发的一项20项患者报告结果(patient-reported outcome,PRO)度量,用于评估疲劳相关症状以及这些症状对RMS患者的生活的影响。②从基线到第108周合并的独特活动性病变(combined unique active lesions,CUAL)的累积数量,时限为从基线至第108周EOT,CUAL定义为通过MRI测量的新的钆增强(Gd+)T1病变脉冲或新的或扩大的T2病变(无重复计数的病变)。③从基线到第108周+30 d研究结束(end of study,EOS)达到确认的残疾累积率(confirmed disability accumulation,CDA)的时间。时限为从基线到第108周+30 d(EOS),相对于研究方案中定义的基线时EDSS评分,为期12周的CDA定义是相对于扩展的EDSS评分而言,扩展EDSS评分有所提高。EDSS得分基于神经科医生的检查,范围为0~10,增量为0.5单位。EDSS可以量化残疾并监控随时间推移残疾水平的变化。④从基线到EOS达到24周CDA的时间,时限为从基线到第108周+30 d(EOS),24周CDA的定义为相对于研究方案中定义的基线时EDSS评分,扩展EDSS评分的增加。EDSS得分基于神经科医生的检查,范围为0~10,增量为0.5单位。EDSS可量化残疾并监控随时间推移残疾水平的变化。
3.2临床试验 代号Optimum,临床试验编号NCT02425644,是一项多中心、随机双盲、平行组的Ⅲ期临床试验,对RMS患者与阳性对照药特氟米特比较疾病控制优势的研究。招募受试者1468例,经筛查,入选1133例,其扩展EDSS在基线时得分为0~5.5分,至少经历过前1年内复发1次,或前2年内复发2次,或至少复发1次。在前6个月或之前6个月脑MRI上发现钆离子增强病变基线。排除原发性进行性MS患者。入选1133例受试者按1:1随机分为两组,A组(n=567)口服庞西莫德片20 mg,qd;B组(n=566)口服特氟米特14 mg,qd,共治疗108周。受试者按照方案完成研究[1,4-5]。
3.2.1受试者人口学与疾病基线特征 可评价的病例数:A组(n=567),B组(n=566),依次顺序列举如下:女性为363例(64.0%)和372例(65.7%);年龄均值与标准差(standard deviation,SD)为(36.7±8.74)岁和(36.8±8.74)岁;基线扩展EDSS评分>3.5分为94例(16.6%)和95例(16.8%)。2年前接受随机疾病修饰治疗为213例(37.6%)和211例(37.3%);前2年根据基线扩大EDSS得分随机接受疾病修饰治疗评分为(2.57±1.17)分和(2.56±1.23)分;研究1年前发生复发受试者为(1.2±0.61)例和(1.3±0.65)例。MS复发缓解亚型为552例(97.4%)和552例(97.5%);继发进展型为15例(2.6%)和14例(2.5%)。基线疲乏症状评分为(31.9±20.4)分和(32.8±19.1)分。基线钆离子增强存在T1病变为226例(39.9%)和256例(45.4%);T2加权病灶体积分别为(8 301.4±10 346.28) mm3和(9 489.2±11 265.42) mm3。高活性病变为202例(35.6%)和200例(35.3%)。
3.2.2主要观察指标疗效评价 可评价的病例数:A组(n=567),B组(n=566),年度主要疗效终点是根据从随机化到EOS或EOT加随访时间,确认每例患者每年复发次数的ARR。EDSS分数由独立、经过培训和认证的评估员使用标准化神经状态的EDSS测试进行评估确诊复发率。ARR:A组242例(42.7%),B组344例(60.8%),A组与B组比较,使ARR降低30.5%(A组平均 ARR减少0.202,B组均值减少0.290;相对危险度(relative risk,RR)为0.695,99%CI=(0.536,0.902),P<0.001,差异有统计学意义。对增强病变存在进行调整的敏感性分析,显示一致的结果。
3.2.3次要观察指标疗效评价 ①FSIQ-RMS,可评价例数:A组(n=449),B组(n=458),各组于每周症状评分,从基线到第108周的变化较低(评分越高表示疲劳度更大)。用最小二乘法计算均值,A组为-0.01,95%CI=(-1.60,1.58),B组为3.56,95%CI=(1.96,5.16),风险比(hazard ratio,HR)=-3.57,95%CI=(-5.83,-1.32),P=0.002。②平均累积组合唯一活跃病灶(mean cumulative combined unique active lesions,MC-CUAL),可评价的病例数:A组(n=539),B组(n=536),A组为1.405,95%CI=(1.215,1.624),B组为3.164,95%CI=(0.316,0.540),HR=0.444,95%CI=(0.36,0.54),P<0.001。③前12周CDA,可评价的病例数:A组(n=567),B组(n=566),A组为57例(10.1%),B组为70例(12.4%),HR=0.83,95%CI=(0.58,1.18),P=0.29。
3.2.4其他探索性观察指标疗效评价 ①患者第一个24周确诊伤残累积病例数,可评价受试者:A组(n=567),B组(n=566)。A组为46例(8.1%),B组为56例(9.9%),HR=0.84,95%CI=(0.57,1.24),P=0.37。②钆增强扫描T1脑高信号病变累计病例数均值,可评价的受试者:A组(n=540),B组(n=538)。A组为0.18,95%CI=(0.141,0.224),B组为0.43,95%CI(0.351,0.525),HR=0.42,95%CI=(0.31,0.56),P<0.001。③用LS计算脑体积变化均值,可评价病例数,A组(n=436),B组(n=434),A组为-0.91,95%CI=(-1.03,-0.79),B组为-1.25,95%CI=(-1.36,-1.13),HR=0.34,95%CI=(0.17,0.50),P<0.001。④评估无复发、无确诊疾病恶化及无新的或扩大的MRI病变等NEDA-3评分均值,可评价病例数,A组(n=564),B组(n=558),A组为25.0,95%CI=(21.4,29.0),B组为16.4,95%CI=(13.5,19.8),HR=1.70,95%CI=(1.27,2.28),P<0.001。⑤评估无疾病活动证据、复发、MRI病变、脑萎缩和残疾进展等4个不同标准的(NEDA-4)评分均值,可评价病例数,A组(n=526),B组(n=532),A组为11.4,95%CI=(8.7,14.6),B组为6.5,95%CI=(4.7,9.0),HR=1.85,95%CI=(1.24,2.76),P=0.003。
4 不良反应概况
开发公司仅公开报道一项代号Optimum、临床试验编号为NCT02425644的多中心、随机双盲、平行组的Ⅲ期临床试验,对RMS患者与阳性对照药特立氟胺比较疾病控制优势的研究。所出现的不良事件列举如下[1,4-5]。
可评价的病例数:A组(n=565),B组(n=566)。①治疗中出现的不良事件(treatment emergent adverse events,TEAEs)分别为502例(88.8%)和499例(88.2%);按系统器官类条款任何一组≥1 TEAEs使治疗中断为49例(8.7%)和34例(6.0%)。②调查发现的不良事件:分别为12例(2.1%)和10例(1.8%);呼吸、胸腔和纵膈障碍为7例(1.2%)和不适用(not applicable,NA);眼疾为5例(0.9%)和NA;胃肠道疾病为4例(0.7%)和4例(0.7%);血液与淋巴系统疾病为3例(0.5%)和2例(0.4%);一般疾病和服药场所条件不适为3例(0.5%)和2例(0.4%);肝胆疾病为3例(0.5%)和2例(0.4%);不适应妊娠期、产褥期和围产期条件为3例(0.5%)和3例(0.5%);血管疾病为3例(0.5%)和NA;神经系统疾病为2例(0.4%)和4例(0.7%);不适应社会环境为2例(0.4%)和1例(0.2%);心脏疾病为1例(0.2%)和2例(0.4%);皮肤和皮下组织功能障碍为1例(0.2%)和2例(0.4%)。③特别关注的不良事件:肝胆疾病或肝功能试验结果异常治疗结束后1 d为128例(22.7%)和69例(12.2%);治疗结束后第15天为145例(25.7%)和82例(14.5%);≥1件严重不良事件为2例(0.4%)和4例(0.7%);高血压为57例(10.1%)和51例(9.0%);肺部事件为45例(8.0%)和15例(2.7%);对心率和心律影响而且第1天低血压为12例(2.1%)和2例(0.4%);疱疹为27例(4.8%)和27例(4.8%);感染为9例(1.6%)和5例(0.9%);癫发作为8例(1.4%)和1例(0.2%);黄斑水肿为6例(1.1%)和1例(0.2%);皮肤恶性状态为5例(0.9%)和1例(0.2%);非皮肤恶性状态为1例(0.2%)和1例(0.2%)。④致命性不良事件为NA和2例(0.4%);严重不良事件(n≥1),按系统器官分组为49例(8.7%)和46例(8.1%);神经系统疾病为9例(1.6%)和6例(1.1%);感染和侵袭为7例(1.2%)和4例(0.7%);胃肠道疾病为6例(1.1%)和4例(0.7%);肿瘤:良性的、恶性的和未指明的,包括囊肿和息肉为6例(1.1%)和3例(0.5%);手术和医疗程序为5例(0.9%)和1例(0.2%);受伤、中毒和手术并发症为4例(0.7%)和7例(1.2%);肾脏和泌尿系统疾病为4例(0.7%)和1例(0.2%);肌肉骨骼和结缔组织组织疾病为3例(0.5%)和4例(0.7%);生殖系统和乳腺疾病为3例(0.5%)和6例(1.1%);肝胆疾病为2例(0.4%)和7例(1.2%);呼吸、胸和纵膈疾病为2例(0.4%)和1例(0.2%);血管疾病为2例(0.4%)和2例(0.4%);血液和淋巴系统疾病为1例(0.2%)和1例(0.2%);精神障碍为1例(0.2%)和2例(0.4%);心脏疾病为0%和2例(0.4%);代谢和营养障碍为0%和1例(0.2%)[1,4-5]。
5 适应证
庞西莫德是一种S1P受体调节药,适用于治疗RMS,包括临床孤立综合征、复发-缓解性疾病和活动性继发性成人进行性疾病[1,4-5]。
6 剂量与服法
6.1剂型与规格 庞西莫德只有速释薄膜包衣片,有2,3,4,5,6,7,8,9,10和20 mg等10种规格[4-5]。
6.2推荐剂量与用法
6.2.1口服首剂庞西莫德片前的检测 ①全血计数(complete blood count,CBC):获取近期,即在过去6个月内或中断先前MS治疗后检测,包括淋巴细胞计数。②心脏评估:获取心电图(electrocardiogram,ECG),确定先前是否存在传导异常;对于有某些既往病史的患者,应听取心脏病专家的建议,寻求首次剂量监测。确定患者是否正在服用减缓心率或房室传导(atrioventricular conduction,AVC)药物。③肝功能检查:获取最近6个月内转氨酶和胆红素水平。④眼科评估:评估眼底,包括黄斑。⑤检查服用影响免疫系统的药物:目前或此前是否服用过对免疫系统有影响的药物,若患者正在接受抗肿瘤、免疫抑制药或免疫调节疗法,或曾经使用过这些药物,考虑可能有非预期口服庞西莫德片添加剂治疗前的免疫抑制作用。⑥接种疫苗:开始口服庞西莫德片前,应检测患者对水痘-带状疱疹病毒(VZV)的抗体;建议对抗体阴性,服用庞西莫德片治疗者应接种疫苗,若接种减毒活疫苗,至少在开始服药前1个月接种疫苗。
6.2.2开始治疗 患者开始口服庞西莫德片,必须使用启动包,开始庞西莫德片治疗,为期14 d的剂量递增服药;第1~2天,每天1次,庞西莫德2 mg;第3~4天,每天1次,庞西莫德3 mg;第5~6天,每天1次,庞西莫德4 mg;第7天,每天1次,庞西莫德5 mg;第8天,每天1次,庞西莫德6 mg;第9天,每天1次,庞西莫德7 mg;第10天,每天1次,庞西莫德8 mg;第11天,每天1次,庞西莫德9 mg;第12~14天,每天1次,庞西莫德10 mg。第15天及以后,按庞西莫德维持剂量,20 mg,每天1次,继续服药。
6.2.3维持剂量 完成剂量递增试验后,从第15天开始,服维持剂量,庞西莫德20 mg,每天1次。药片须整片吞服,是否与食物同服均可。
6.3有一定心脏病史患者口服首剂量的监测条件 因患者给予庞西莫德开始治疗,会导致患者心率下降,首次剂量对窦性心动过缓(心率<55次·min-1)患者及每分钟一级或二级MobitzI型房室传导阻滞,或心肌病史患者,治疗前6个月以上发生梗死或心力衰竭,且病情稳定条件。建议监护4 h[4-5]。
6.3.1首剂4 h的监测 在资源需要适当管理的情况下,服首剂庞西莫德片后,可监测出患者是否有心动过缓。首剂服药后4 h监测患者的症状、心动过缓症状、每小时脉搏和血压最低测量值。在给药前和给药后4 h是对这些患者进行心电图检查的观察期。
6.3.24 h监控后的附加检测 若4 h后出现以下任何异常情况,即使没有症状,必须继续监测,直到异常情况消失:①给药后4 h心率<45次·min-1;②给药后4 h心率处于最低值,表明对心脏的最大药效作用可能尚未发挥;③给药后4 h心电图显示新发二度或更高房室传导阻滞,如给药后出现症状性心动过缓、缓慢心律失常或传导相关症状,或给药后4 h心电图显示新发二度或更高房室传导阻滞或QTc间期比≤500 ms更大,必须开始适当处置,连续监测心电图,若无药物治疗,继续监测直至症状消失。如需药物治疗,继续监测过夜,并重复第2次给药后4 h监测。应根据心脏病专家的建议,确定最适当的监测和治疗开始时的策略,包括夜间监测或考虑服用庞西莫德片;④患有心脏和脑血管疾病患者;⑤给药前或4 h观察期间,或QT间期延长或同时服用延长QT间期药物治疗的额外风险,有点扭转性心动过速的危险;⑥同时接受减慢心率或房室传导的药物治疗。
6.4治疗中断后重新启动庞西莫德片治疗 不建议在治疗期间中断服药,尤其是错过<4次连续剂量;①在剂量递增过程中错过服药,恢复所错过的递增剂量服药,并在该剂量递增日程时间表继续往后日程进行服药;②维持剂量:恢复维持剂量继续治疗。如需要在剂量递增方案第1天重新开始治疗,即新的起始包,必须对患者进行完整的首剂监测[4-5]。
7 禁忌证
禁止下列患者服用庞西莫德片治疗:①在过去的6个月里,经历过心肌梗死、不稳定心绞痛、卒中、短暂性脑缺血发作(transient ischemic attack,TIA),需要住院治疗的失代偿性心力衰竭,或Ⅲ或IV级心力衰竭。②有MobitzⅡ型二度、三度房室传导阻滞,或病态窦房结综合征,或窦房传导阻滞,除非患者有安装功能性起搏器[4-5]。
8 用药注意事项与警示
8.1感染 庞西莫德是S1P受体调节药。可使外周血淋巴细胞计数呈剂量相关性降至30%~40%,可能对病毒感染的敏感性增加,并增加与其他疾病和罕见相关致命性感染。临床研究表明庞西莫德20 mg组(简称A组)与特氟米特14 mg组(简称B组)比较,两组之间感染率有可比性,但严重或重度感染发生率分别为1.6%和0.9%。对于有活动性感染的患者,应推迟庞西莫德片治疗,直至缓解[4-5]。
8.1.1疱疹病毒感染 在庞西莫德发展计划中报告疱疹病毒感染病例;单疱病毒性脑炎、水痘-带状疱疹性脑膜炎在其他S1P受体调节药也有报道,特别是未经疫苗接种的受试者。
8.1.2隐球菌感染 出现致命性隐球菌性脑膜炎(cryptococcal meningitis,CM)和播散性隐球菌感染病例,在其他S1P受体调节药曾有报道。医生应警惕临床症状或迹象。若患者症状或体征与隐球菌感染一致,应及时诊断和对症治疗,并暂停庞西莫德片治疗,若诊断为CM,应及时治疗,痊愈后才能启动庞西莫德片治疗。
8.1.3进行性多病灶脑白质病(progressive multifocal leukoencephalopathy,PML) PML是一种病毒感染的机会性疾病,由詹姆斯敦峡谷病毒(Jamestown Canyon virus,JCV)感染引起的脑部疾病,只发生在免疫功能低下患者,会导致死亡或严重残疾。典型症状为进行性虚弱、身体一侧或四肢笨拙、视觉障碍、思维改变、记忆和取向混乱和性格改变,进展持续数天至数周。患者服用S1P受体调节剂和其他多种药物治疗会出现PML硬化症,治疗与一些危险因素有关,如患者免疫功能低下,使用免疫抑制剂进行综合治疗。医生应警惕可能出现的临床症状或MRI发现暗示PML。若临床症状或体征出现之前,若怀疑PML,在排除PML前,应暂停服庞西莫德片治疗,PML被证实后,终止庞西莫德治疗。
8.1.4抗肿瘤、免疫调节或免疫抑制疗法 抗肿瘤、免疫调节或免疫抑制疗法,包括皮质类固醇,应谨慎联用,有提高免疫系统影响的风险。
8.1.5免疫接种 若不能确认罹患水痘病史或接种过疫苗,庞西莫德片开始治疗前应检测VZV抗体。患者抗体阴性,在治疗前应接种水痘疫苗,将庞西莫德片治疗推迟4周,让疫苗产生效果。目前尚无关于接种疫苗有效性和安全性的临床数据。若在庞西莫德片治疗期间接种疫苗,效果可能会降低。需要接种减毒活疫苗,应在庞西莫德片开始治疗前至少1个月。避免在治疗后1~2周内接种减毒活疫苗。
8.2缓慢心律失常和房室传导延迟 庞西莫德片开始治疗会导致心率短暂下降,房室传导延迟,必须使用剂量递增治疗方案,才能达到庞西莫德维持剂量20 mg的研究。临床试验研究不包括以下患者:①在基线心电图上,静息心率<50次·min-1;②筛查时观察到Mobitz Ⅱ型二级房室传导阻滞或更高级别房室传导阻滞;③心力衰竭(纽约心脏病协会Ⅲ-Ⅳ级)或存在任何严重心脏病;④心脏传导或心律失常(包括有症状的窦房传导阻滞)心动过缓、心房扑动或心房颤动、室性心律失常、心脏骤停)病史或筛查时观察到的病情;⑤筛查时观察到MobitzⅡ型二级房室传导阻滞或更高级别房室传导阻滞;⑥筛查时QTcF间期女性>470 ms,而男性>450 ms;⑦有与心脏疾病相关的晕厥病史;⑧全身动脉高血压失控;⑨与心脏疾病相关的晕厥病史;⑩全身动脉高血压失控[4-5]。
8.2.1心率减慢 开始庞西莫德片治疗可能导致心率短暂下降。在研究1中,在治疗开始,心电图窦性心动过缓,A组发生率为5.8%,而B组为1.6%。在首次剂量递增服药后,心率通常在1 h内开始下降,并在2~4 h内达到最低点。心率通常给药后4~5 h后恢复到基线水平。第1天的心率平均下降6次·min-1。给药后第1天进行剂量递增,心率下降较不明显。在研究1中,所有心动过缓患者在未经干预的情况下均得到缓解,并未发生要求停服庞西莫德片。第1天,3例患者接受庞西莫德片治疗,给药后心率≤40次·min-1;这3例患者都有基线心率<55次·min-1。
8.2.2房室传导延迟 开始庞西莫德片治疗与短暂性房室传导延缓有关,并与剂量递增期间的心率下降相似模式延迟。在研究1,A组房室传导延迟表现为一级房室传导阻滞,延长心电图PR间期的发生率为3.4%,而B组为1.2%。传导异常通常是短暂的,无症状,24 h内解决,在研究中,曾对二级和三级房室传导阻滞进行研究,接受庞西莫德片治疗的患者未出现过,患者若需要接受治疗,可寻求心脏病专家的建议:①QT明显延长>500 ms;②对心房扑动/颤动或心律失常患者应服用Ⅰa类或Ⅲ类抗心律失常药物治疗;③患有不稳定型缺血性心脏病,心脏失代偿性心力衰竭发生率超过治疗开始前6个月,有心脏骤停史,短暂性脑缺血发作,中风发生在治疗开始前6个月以上的脑血管疾病或未控制高血压;④有MobitzⅡ型≥二级房室传导阻滞史,患有病态窦房结综合征或窦房心传导阻滞者。
8.2.3启动治疗的建议 ①所有患者均应获得心电图,以确定目前是否存在传导异常;②建议采用剂量梯度递增法开始治疗,以降低心脏效应;③患者有窦性心动过缓,一级或二级(Mobitz I型)房室传导阻滞,或术前>6个月有心肌梗死或心力衰竭病史,开始治疗时,建议服首次剂量进行监测;④有心搏、脑血管病史患者不推荐使用庞西莫德片治疗下列疾病:TIA、治疗开始前>6个月发生卒中,因明显心动过缓,高血压或严重未经治疗的睡眠呼吸暂停。此类患者可能耐受性差,若考虑治疗,在开始治疗前,应寻求心脏病学家意见,确定最有效的适当监测策略;⑤有反复晕厥或症状性晕厥病史患者服用庞西莫德片,应对于心动过缓的总体获益风险进行评估。从治疗有效性考虑,治疗前应咨询心脏病专家意见,确定最合适的监控:⑥对同时服治疗药物的患者,如降低心率的β受体阻断药、非二氢吡啶钙通道阻滞药地尔硫和维拉帕米和地高辛等同时与庞西莫德片服用,可能与发生严重的疾病有关,会引发心动过缓和心脏传导阻滞。在开始治疗前应寻求心脏病专家建议,确定最适宜的监测方法;⑦患者接受稳定剂量β受体阻断药,若静息心率>55次·min-1,允许加服庞西莫德片,如静息心率≤55次·min-1,应暂停β受体阻断药治疗直至基线心率>55次·min-1,可恢复庞西莫德片加服β受体阻断药治疗;⑧患者在服庞西莫德片治疗期间,加服其他降低心率药物,对心率有潜在的附加效应,应咨询心血管疾病专家的意见。
8.2.4治疗开始或维持治疗期间遗漏剂量 若在治疗开始或维持期间连续错过≥4次每日治疗剂量,重新开始第1天的梯度递增剂量(用新的启动包),并遵循第一剂量监测建议。
8.3影响呼吸 在研究1中,患者FEV1减少与剂量呈正相关,一氧化碳弥散量(carbon monoxide diffusing capacity,DLCO)减少多数发生在治疗开始后第1个月。A组治疗2年,预测FEV1减少8.3%,有7例患者因为肺部不良反应而停药。而B组为4.4%。因信息不足,无法确定停药后FEV1或FVC下降的可逆性。庞西莫德对患有肺纤维化、哮喘和慢性阻塞性肺疾病等严重呼吸系统疾病有影响。若有临床指征,应在治疗期间进行处置[4-5]。
8.4肝损伤 患者经庞西莫德片治疗可能发生转氨酶升高。临床研究1,A组丙氨酸氨基转移酶(ALT)升高≥5倍正常上限(ULN)为4.6%,B组为2.5%;ALT升高≥3倍ULN两组分别为17.3%和8.3%。所有ALT升高与服药时间呈正相关。ALT升高3倍的中位时间为3个月,2~4周内恢复至 8.5血压升高 A组收缩压均值升高2.9 mmHg(1 mmHg=0.133 kPa),舒张压升高2.8 mmHg;B组分别升高2.8和31 mmHg。血压升高约治疗后1个月出现,继续治疗,两组报告高血压不良反应分别为10.1%和9.0%。1例患者报告出现高血压危象。治疗期间应监测高血压性心脏病[4-5]。 8.6皮肤恶性肿瘤 临床研究曾报道S1P受体调节药,包括庞西莫德片会出现基底细胞癌和其他皮肤恶性肿瘤的病例。基底细胞癌的发生率,A组为0.4%,B组为0.2%;其他皮肤恶性肿瘤,包括黑色素瘤和鳞状细胞癌,用其他S1P调节剂治疗也有报道。建议患者定期进行皮肤检查,尤其是有皮肤病危险因素的皮肤癌患者。若发现可疑皮损,应及时评估。应限制暴露在阳光和紫外线下,需穿着防护服,用高防护系数的防晒霜。接受庞西莫德片治疗的患者不建议用UV-B辐射或光化学疗法,将增加患皮肤癌的风险[4-5]。 8.7黄斑水肿 S1P受体调节剂,包括庞西莫德,会增加黄斑水肿的发病率。临床研究表明,A组出现黄斑水肿为1.1%,B组为0%。在开始治疗之前,所有患者都应对包括黄斑的眼底进行眼科检查,若患者在治疗期间报告视力有任何变化,可随时再次进行检查和处置。有葡萄膜炎或糖尿病史的患者,曾罹患黄斑水肿,S1P受体调节剂治疗期间的黄斑水肿的发病率增加,应定期随访,检查眼底。 8.8后侧可逆脑病综合征(posterior reversible ence-phalopathy syndrome,PRES) PRES的病例较少见。接受S1P受体调节剂治疗的患者尚未发现这样的病例。A组经治疗的患者若出现任何意想不到的神经或精神症状/体征,如认知缺陷,行为改变,皮质视觉障碍或其他神经皮质症状/体征,应及时诊治。PRES的症状通常是可逆的,但可能演变为缺血性中风或脑卒中出血。延误诊断和治疗可能导致永久性的神经后遗症。如怀疑是PRES,应停止给予庞西莫德片[4-5]。 8.9此前免疫抑制药治疗的非预期遗留免疫抑制作用 用免疫抑制或免疫调节疗法当从具有长期免疫效果的药物转换时,必须考虑这些药物的半衰期和作用方式,避免对免疫系统产生意外的附加效应,最大限度地降低风险性疾病重新激活时,启动庞西莫德片治疗。不建议在用阿来单抗治疗后开始用庞西莫德片治疗[4-5]。 8.10停止庞西莫德片用药后使残疾严重性增加 严重恶化疾病,包括疾病反弹,很少报道停服S1P受体调节药后疾病严重恶化的可能性。应在停服庞西莫德片治疗后考虑观察患者在停药后残疾严重性增加,并应进行适当的治疗处置。 8.11停止庞西莫德片用药后对免疫系统的影响 停服庞西莫德片后,血液中还滞留长达约1周。在此期间内开始其他疗法,将导致伴随接触庞西莫德效应。90%患者在停药后1周内淋巴细胞计数恢复正常,模型研究显示,庞西莫德剩余药效学效应,如降低外周血淋巴细胞计数,可能持续存在于末次给药后1~2周。此期间服用免疫抑制药,可能导致对免疫系统产生附加效应,应谨慎等待在末次停止服庞西莫德1~2周后再更换新治疗方案[4-5]。 8.12胎儿风险与妊娠妇女用药 目前尚无关于妊娠妇女口服庞西莫德片的充分及良好对照研究数据。动物实验研究表明,妊娠期间喂饲庞西莫德对动物产生不良影响,包括胚胎死亡率升高和胎儿畸形毒性。孕大鼠和孕兔在胚胎发育期,接触庞西莫德,对内脏和骨骼畸形发生很大影响。孕大鼠在器官形成期,喂饲庞西莫德0,1,10或40 mg·kg-1·d-1,除最小剂量组外,其余剂量组均观察到器官缺陷,主要累及胎儿,增加畸形发育,四肢并指、直肠和心血管系统,包括室间隔,胚胎和胎鼠死亡率高。未观察到母体毒性,表明对胎鼠的选择性作用。无毒剂量为1 mg·kg-1·d-1,血浆接触量(AUC)对大鼠胚胎-胎鼠发育的不良影响低于RHD20 mg·d-1[4-5]。 8.13哺乳期妇女用药 尚无关于母乳汁中是否存在庞西莫德,也无关于庞西莫德对母乳喂养婴儿的影响或药物对分泌乳汁的影响数据。给哺乳期雌大鼠喂饲庞西莫德,其子代血浆可检测到庞西莫德,提示庞西莫德可排泄入乳汁中。哺乳期妇女在服庞西莫德片治疗期间是否母乳喂养婴儿应权衡对婴儿发育和健康的好处,母亲临床治疗需求及母乳喂养婴儿的任何潜在不利影响或庞西莫德潜在的不良反应[4-5]。 8.14有生殖潜力的男女伴侣用药 开始给予庞西莫德片治疗前,女性应咨询了解该药对胎儿可能有严重危害及在怀孕期间采取有效避孕措施必要性;用庞西莫德片治疗后,需要在停服末次剂量约1周后,使体内清除残留的庞西莫德,对患者体内潜在的胎儿危险性可能持续存在,在此期间,女性及其男性伴侣需采取有效的避孕措施[4-5]。 8.15儿童患者用药 对儿童用药其安全性和有效性尚未确定。幼龄动物毒性研究表明,对出生后第2~91天幼大鼠喂饲庞西莫德0,1,10,30或100 mg·kg-1·d-1,测试两个最在剂量,观察到肺组织病理学改变,肺泡组织细胞增生和(或)水肿,并减少免疫功能的T细胞依赖性抗体反应。体质量增加和(或)长骨长度生长速度缓慢,但测试低于2个最大剂量组,观察到神经行为损伤,运动活动增加。在实验结束后,经为期4周恢复期,淋巴细胞计数下降和神经行为损伤仍持续存在[4-5]。 8.16老年患者用药 庞西莫德片的临床研究不包括年龄≥65岁老年患者。老年患者的反应不同于年轻人。老年患者应慎用庞西莫德片,因老年患者的肝、肾或心脏功能下降的概率更高,且经常伴随疾病或服用其他药物治疗[4-5]。 8.17肝损伤患者用药 轻度肝损伤(Child-Pugh A级)患者无需调整服药剂量。对于中度或重度肝损害患者(Child-Pugh B级及C级),不建议服用庞西莫德片,因不良反应的风险可能更大[4-5]。 美国FDA给予研发公司排他性保护期至2026年3月18日期满;研发公司申请3份品种保护专利:US8273779、US9000018和USRE43728均已授权,保护期至2024年11月16日期满,相应中国专利CN1882555也已授权,专利期与美国同日期满。该公司申请一份保护晶型的美国专利:US9062014,已获授权,专利期至2032年5月6日期满,相应中国专利CN102177144也已授权,专利期至2029年10月19日期满。研发公司申请一份美国专利,保护S1P1受体激动剂的给药方案专利,已授权,专利期至2035年12月10日期满,相应中国专利CN106999462已授权,专利期与美国同日期满,分案申请专利CN106999461尚在实审中。笔者尚未查阅到研发公司向国家药品监督管理局提出申请进口注册证的信息。9 知识产权状态与国内外研究进展
