新型致癌蛋白:Gankyrin
2021-10-06谢芳芳陈杰胡蝶黄德发钟田雨
谢芳芳,陈杰,胡蝶,黄德发,钟田雨,2
(1.赣南医学院第一临床医学院,江西 赣州 341000;2.赣南医学院第一附属医院精准医学中心,江西 赣州 341000)
癌症是体内异常细胞出现异常增殖并侵袭临近部位和扩散到其他器官的现象。截止至2018年全球癌症亡人数将达到990 万人[1]。以肺癌、肝癌、结直肠癌、胃癌和乳腺癌为主。早期诊断可以提高癌症的治愈率以及肿瘤患者的生存时间,因此寻找和发现早期诊断标志物在癌症早期诊断中至关重要。研究发现gankyrin在许多肿瘤中存在高表达现象,并且其表达水平与肿瘤的进展密切相关[2,3]。与低表达或不表达gankyrin的肿瘤相比,高表达gankyrin的肿瘤恶性程度较高且与组织学分级相关[4-6]。因此,可以将gankyrin 作为早期诊断和评估癌症恶性程度的指标。肿瘤细胞中高水平Gankyrin一定程度上与肿瘤的转移和不良预后密切相关[7],同时gankyrin 还参与肿瘤耐药表型的形成[8-10]。因此,靶向gankyrin 疗法将成为临床上治疗化疗耐药患者新的有效手段。
1 Gankyrin与癌症
Gankyrin基因编码的Gankyrin 又称PSMD10,是一种由7 个锚蛋白重复序列(ARs)组成的重复连接蛋白。该序列最初发现于人连接蛋白中,参与蛋白-蛋白相互作用,在细胞周期进展、凋亡和肿瘤发生中发挥重要作用。作为蛋白酶体复合体的亚基,gankyrin通过与26S蛋白酶体中的S6b ATP酶相互作用,实现蛋白酶体介导的降解[11-13]。
Gankyrin 最初发现于原发性肝癌(HCC),且在正常组织和癌组织间存在差异表达[14],被认为是H CC 病理诊断和肝癌发生的重要生物学标志物。Gankyrin通过调控肿瘤抑制蛋白P53和pRB 活性,打破肿瘤细胞的增殖凋亡稳态,发挥促癌作用[15]。TP53和RB1 抑癌基因是肿瘤发生过程中常见的突变靶点。TP53 编码的P53通过诱导细胞周期阻滞和细胞凋亡,下调癌基因表达;视网膜母细胞瘤蛋白(pRB)则通过调节细胞凋亡,维持细胞生长与凋亡的稳态[16]。P53和pRB 两者协调共同维持机体稳态。Gankyrin的C 端环指结构域通过结合E3泛素连接酶MDM2 增加P53 泛素化和降解[17],178LxCxE182 保守基序结合pRB 并诱导其磷酸化后经蛋白酶体途径降解[18,19],最终导致细胞增殖失控,引发恶性肿瘤(图1)。Gankyrin 不仅能够加速癌细胞的增殖和扩散,促进癌变和转移[20-22];同时可以作为非小细胞型肺癌p-TNM 分期和淋巴转移的预后因素[23,24]。Gankyrin在多种肿瘤中高表达的特性以及高相关性使其越来越受研究人员的关注。

图1 Cankyrin通过调控P53和pRB 活性促进癌细胞增殖。A.DNA 损伤后,p53 阳碍细胞周期的进展,迫使细胞停留在G1 阻滞状态,直到DNA 得以修复;当损伤范围扩大,细胞就会进入凋亡状态。Gankyrin通过MDM2和26S蛋白酶体的S6B 亚基结合,将泛素化的p53 传递到蛋白酶体降解,P53 经降解后癌细胞从G1阻滞状态进入细胞周期的S 期。B.Gankyrin与pRB 结合并使其发生磷酸化,随后经蛋白酶体途径降解。pRB的降解会使E2F(一种激活细胞周期蛋白依赖性激酶(CDKs)的转录因子)释放。后者激活CDKs,导致癌结胞进入结胞周期的S 期。
2 Gankyrin 促进肿瘤细胞增殖、转移和耐药
Gankyrin通过激活PI3K/AKT/HIF-1α通路促进肝癌发展[14]。PI3K/AKT 信号通路是细胞调节网络的核心,影响细胞的生长、分化和代谢。Gankyrin调节PI3K/AKT通路,激活HIF-1α,从而调控twist相关蛋白(Twist1)、血管内皮生长因子(VEGF)和基质金属蛋白酶2(MMP-2)等肿瘤增殖和转移相关蛋白的表达[25]。过表达的gankyrin 上调胰岛素样生长因子结合蛋白5(IGFBP-5)促进Huh-7 肝癌细胞的增殖[26]。此外还包括上调Ras 同源基因家族成员A(RhoA)抑制PTEN 增加PIP3 磷酸化。活化的PI3K/AKT 信号通路,进一步增强gankyrin和cyclin D1的表达。该机制已经在子宫内膜癌和卵巢癌中得到了验证[27,28]。值得注意的是,下调gankyrin 除了抑制癌细胞的生长和增殖外,还能增强肿瘤细胞对化疗药物的敏感性(详见下文),可作为临床治疗肿瘤的潜在靶点[29]。
Wnt/β-catenin通路是一种保守的信号通路,在调节细胞生长、增殖和肿瘤发生中发挥重要作用[30,31]。其中β-连环蛋白(β-catenin)通过调控c-Myc和cyclin D1 调节细胞生长,糖原合成酶激酶-3(GSK3)辅助过量的β-catenin 降解以保证其稳态。过度激活的Wnt 信号抑制GSK3,导致βcatenin 积累,最终引发细胞增殖失控[32]。一项关于HCC的研究发现,gankyrin 能够上调c-Myc 影响肝癌细胞的增殖,增强肝癌细胞的糖酵解和谷氨酰胺的分解,促进肝癌的发生、转移和耐药[33]。Gankyrin与β-catenin 存在正反馈回路,gankyrin上调β-catenin 转录活性使β-catenin的表达增加;同时过表达的β-catenin 也增强了gankyrin的表达,两者相互作用,促进肿瘤的发生和转移[34]。Gankyrin与IL-6/STAT3 信号通路也存在正反馈激活。Gankyrin通过促进RB 磷酸化调节IL-6的表达,同时IL-6 经IL-6/STAT3 正反馈上调gankyrin表达,促进cyclin D1和VEGF以及MMP-2和MMP-9表达,促进癌细胞的增殖和血管的生成[35]。同样的作用在非小细胞型肺癌上皮-间质转化上也得到了证实,提示gankyrin 可作为临床肿瘤恶性程度的参考指标[36]。
核转录因子ReIA/NF-κB 控制机体许多重要的生物学功能,包括DNA 转录、细胞增殖及凋亡[37]。NF-κB与细胞的凋亡密切相关,通过调节促凋亡基因和抗凋亡基因的表达,影响肿瘤细胞凋亡[38]。肿瘤细胞中过度活化的NF-κB 激活下游基因CyclinD1和c-Myc 刺激细胞生长,导致增殖失控;同时NF-κB 能够促进肿瘤转移相关基因ICAM-1、MMP-9 等的表达,加速肿瘤的转移。然而在正常肝细胞中,低活性的NF-κB 抑制其下游的抗凋亡基因的表达,促进肿瘤的产生。肝细胞中高表达的gnakyrin与ReIA 结合并通过SIRT1调节ReIA 乙酰化,从而抑制NF-κB 转录活性[39]。Gankyrin的上述致瘤作用可能与减弱NF-κB 抑制抗凋亡基因的表达有关,包括MDM2 介导的泛素化和蛋白酶体介导的p53 降解的增加,通过抑制NF-κB 活性,进一步促进肝癌的发生[17,40]。
Gankyrin 参与肿瘤细胞的增殖侵袭和转移,并且与肿瘤细胞的化疗敏感性密切相关。传统化疗药物诱导的细胞凋亡主要是通过TP53 依赖途径介导的[41]。研究发现HCC 耐药的可能与TP53基因的缺陷或者丢失相关[42]。抑制gankyrin 介导的P53泛素化降解,可以从根源提高肿瘤细胞的凋亡水平,实现抗癌药物敏感化[43]。下调的gankyrin 可以增强胃癌细胞对5-氟尿嘧啶和顺铂的化疗敏感性[44]。此外,gankyrin 能够结合热休克转录因子(HSF1)促进ATG7表达,增强自噬通量以抵抗HCC患者索拉非尼治疗[45];抑制gankyrin 介导的P53 泛素化可以改善肿瘤细胞化疗不敏感性[19]。有趣的是,研究人员发现在TP53 阴性的肝癌细胞株Hep3B 中同样存在敲低gankyrin 诱导肿瘤细胞凋亡的现象。表明gankyrin 调节肿瘤细胞凋亡存在不依赖P53的通路[46]。Gankyrin 调节肿瘤细胞凋亡的相关通路还有待进行深入的研究。
3 基于Gankyrin蛋白的肿瘤治疗
一项关于gankyrin与肿瘤预后相关性的荟萃分析结果显示gankyrin 可作为新的预测生物标志物和新型治疗靶标[21,47-49]。该结果引起了研究者对“gankyrin 抑制”治疗的兴趣,即对gankyrin基因进行不同水平的调控,包括基因水平、转录水平、翻译水平、和翻译后水平的调控实现gankyrin 抑制,改善肿瘤预后。
小干扰RNA(siRNAs)又称作非编码双链RNA,通过RNA 干扰机制特异性抑制mRNA表达[50,51]。利用siRNA 下调gankyrin表达,通过诱导pRB 去磷酸化抑制细胞周期G1-S 期相关基因表达,使肝癌细胞增殖受阻,从而抑制肿瘤细胞的生长[43]。Gankyrin的转录后水平受miRNA 调控。miR-214通过下调gankyrin 抑制骨髓瘤细胞生长[52]。miR-605 可直接结合gankyrin的3’UTR 区域,降低其在肝内胆管癌(ICC)中的表达,通过P53/miR-605/PSMD10/MDM2 正反馈环抑制ICC细胞的增殖和侵袭[53]。miR-532-3p通过Gankyrin/AKT 信号通路调控肝细胞癌的上皮-间充质转化和转移[54]。基于miRNA 转录后水平调控gankyrin表达对肿瘤表型影响的结果表明,下调gankyrin 可以作为临床上治疗癌症的有效手段,研究和开发能够结合gankyrin 并抑制与之相关的促癌通路成为今后研究的重点。
表观遗传的异常改变在gankyrin 相关肿瘤的治疗方面越来越受重视。甲基化和组蛋白修饰是两种最常见的表观遗传修饰,通过影响相关基因的转录因子来调节该基因的表达[12]。长链非编码RNA Linc-GALH通过泛素化降解DNA 甲基转移酶(DNMTs),使gankyrin 发生去甲基化[55],组蛋白去乙酰化酶(HDAC)通过去除组蛋白上的乙酰基,改变染色质结构构象使转录失活[56]。研究发现组蛋白去乙酰化酶(HDAC)在调节gankyrin表达方面存在多效应。广谱组蛋白去乙酰化酶抑制剂LBH589通过阻断Gankyrin/STAT3/AKT 信号通路,改善HCCs的增殖和转移[57];然而,上调的HDAC同样可以和gankyrin 启动子区C/EBPb 转录因子结合,通过抑制C/EBP 依赖性启动子发挥下调gankyrin表达的作用[58]。综上,HDAC 可作为靶向gankyrin 治疗癌症的潜在靶点。
最近的研究侧重于gankyrin蛋白表面的蛋白-蛋白相互作用(protein-protein interactions,PPI)。Gankyrin PPI 可以调节机体的肿瘤抑制蛋白P53和pRB,从而实现gankyrin的促癌作用。因此,破坏或抑制gankyrin蛋白表面PPIs 成为了gankyrin靶向治疗癌症的新策略。已有相关研究证明小分子合成蛋白Gankyrin binding protein (GBPs)能够通过靶向gankyrin的PPI,调节gankyrin与S6 ATP 酶之间的相互作用,从而抑制Gankyrin/MDM2依赖的P53 泛素化[59]。同样的作用在小分子cjoc42中也得到了证实[60]。虽然关于gankyrin PPI 抑制剂的研究已经取得的了初步的进展,但是目前尚未发现能够特异性结合gankyrin的PPI 抑制剂,该领域还有待进行深入的研究。
4 总结
Gankyrin 作为新型致癌蛋白,被认为是许多肿瘤的生物标志物[61]。Gankyrin通过调节肿瘤细胞内抑癌基因P53和pRB的活性和数量,导致肿瘤细胞过度增殖,引发恶性肿瘤。除此之外,gankyrin 还能调节肿瘤细胞生长、增殖相关的信号通路,包括PI3K/AKT信号通路、Wnt/β-catenin通路、IL-6/STAT3、核转录因子ReIA/NF-κB 等促进肿瘤细胞的增殖、侵袭和转移。降低肿瘤细胞gankyrin水平能够增强肿瘤细胞对化疗药物的敏感性,其作用机制包括gankyrin 介导的P53 降解减弱和自噬抑制。综上,肿瘤组织中特异性表达的gankyrin 不仅可以作为临床上多种肿瘤的诊断标志,同时还能作为治疗和改善肿瘤化疗耐药的潜在靶点,见图2。利用小干扰RNA(siRNA)、表观遗传修饰、转录后水平的调控等方式调节机体内gankyrin水平以及破坏gankyrin表面PPI 从而降低其生物学活性达到抑制肿瘤的作用,成为了gankyrin靶向治疗的重要手段。小分子合成蛋白(GBPs)和cjoc42 正是基于上述作用机制实现肿瘤的治疗。虽然关于gankyrin PPI的研究已经有了很大的收获,但是尚未发现直接的gankyrin 结合物能够调节癌细胞内的gankyrin蛋白,在这方面,仍然存在很大的空间和挑战。
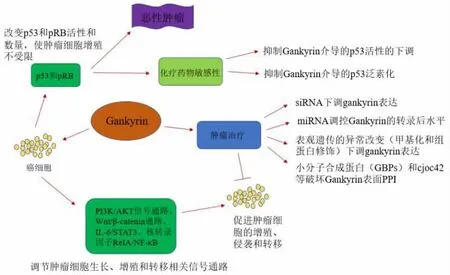
图2 Gankyrin 相关功能模式图
