代谢工程改造大肠杆菌生产L-丝氨酸
2021-09-18李旋王加初刘益宁蒋帅吴鹤云谢希贤
李旋,王加初,刘益宁,蒋帅,吴鹤云,谢希贤,3*
1(天津科技大学 生物工程学院,天津,300457) 2(天津科技大学 食品科学与工程学院,天津,300457) 3(代谢控制发酵技术国家地方联合工程实验室,天津,300457)
L-丝氨酸(L-serine,L-Ser)可以促进脂肪以及脂肪酸的合成,同时还是半胱氨酸和色氨酸等物质的合成前体,具有重要的生理功能[1],被广泛应用于食品、医药和化妆品等领域。目前L-Ser的生产方法主要包括化学合成法、酶催化法和微生物发酵法,其中微生物发酵法原料来源广泛,能耗低,对环境友好,是L-Ser生产研究的重点。
生物体中L-Ser合成途径受到严谨的调控,且生成的L-Ser很容易被降解(图1)。另外,胞内L-Ser积累不仅会抑制细胞分裂和肽聚糖的合成[2],还会转化为丙烯酸酯等有毒物质对细胞的生长产生抑制[3]。这些因素限制了L-Ser的过量合成。因此,L-Ser高产菌株的构建策略主要包括3个方面:(1)解除反馈调控作用以增强L-Ser合成通量。BELL等[4]通过去除3-磷酸甘油脱氢酶(PGDH,serA基因编码)的调控域,所得PGDH突变体不受L-Ser反馈抑制,有效促进了L-Ser的合成。(2)阻断L-Ser降解的途径。L-Ser可被降解为丙酮酸和甘氨酸,直接阻断L-Ser向丙酮酸的降解有利于L-Ser的积累,同时不影响细胞正常代谢[5];但是L-Ser向甘氨酸裂解过程中产生的5,10-亚甲基四氢叶酸对于嘌呤的合成有着关键作用,直接阻断此途径会对菌体生长产生严重的影响。STOLZ等[6]为了弱化L-Ser向甘氨酸的降解,敲除了谷氨酸棒状杆菌中pabAB和pabC基因,构建了叶酸缺陷性菌株,经过60 h发酵,可积累L-Ser约36 g/L。但是在发酵过程中需添加适量叶酸来调控丝氨酸羟甲基转移酶(SHMT,glyA基因编码)的活性,并且生长也受到明显抑制。(3)提高细胞对L-Ser的耐受性。一是通过适应性进化的方式逐步提升菌株对L-Ser的耐受性,如MUNDHADA等[7]对菌株进行适应性进化,提高了菌株对高浓度L-Ser的耐受性,最终耐受菌经过48 h补料分批发酵,产物积累量为37 g/L,L-Ser产量提高了3倍,对进化菌株进行全基因组测序发现thrA基因突变对减缓L-Ser对细胞毒性有重要的作用。二是选择合适的转运蛋白促进胞内L-Ser分泌,从而减缓胞内L-Ser积累对菌体的生长抑制。ZHANG等[8]通过对基因eamA进行同源检索,发现谷氨酸棒状杆菌中蛋白SerE可高效转运L-Ser,菌株经过118 h补料分批发酵,终产量达到50 g/L,这是目前报道的L-丝氨酸的最高产量。
目前,L-Ser菌株构建取得了一定的成效,但是一些关键问题如L-Ser向甘氨酸的降解以及菌株对L-Ser的耐受性等还未得到很好的解决。为此本研究以野生型大肠杆菌MG1655为出发菌,通过减弱L-Ser降解途径,增强L-Ser的合成酶类的表达,以及改造L-Ser的转运系统,获得了1株稳定高产,无质粒的非缺陷型大肠杆菌L-Ser生产菌株(图1)。其中,为了减弱丝氨酸向甘氨酸的降解,采用启动子Ptrp启动glyA基因的转录表达,通过调整色氨酸的添加时间和添加量对SHMT的活性进行动态调控,有效促进了L-Ser的积累,且未对菌体生长产生明显影响,可为L-Ser高产菌株的构建提供新的借鉴和参考。

图1 L-Ser生物合成途径和本研究的代谢工程改造策略Fig.1 Biosynthetic pathway of L-Ser and the metabolic engineering strategies used in this study
1 材料与方法
1.1 菌株、质粒以及主要试剂
实验所用菌株及质粒见表1。Primer STAR HS DNA聚合酶,大连宝生物科技有限公司;2×Rapid Taq Mix、ClonExpress®II One Step Cloning Kit,南京诺唯赞生物科技有限公司;质粒快速提取试剂盒、DNA凝胶纯化回收试剂盒,美国Omega公司;引物及基因均由苏州金唯智科技有限公司合成。

表1 实验所用菌株和质粒Table 1 Strains and plasmids used in this study
1.2 工程菌株构建
根据LI等[9]报道的CRISPR/Cas9基因编辑技术对E.coli基因组进行编辑。操作体系包含pGRB和pREDCas9这2个质粒。其中pREDCas9带有gRNA质粒的消除系统,λ噬菌体的Red重组系统及Cas9蛋白表达系统,拥有奇霉素抗性,32 ℃培养;pGRB质粒是以pUC18为骨架,质粒含有启动子J23100,gRNA-Cas9结合区域序列和终止子序列,具有氨苄青霉素抗性,37 ℃培养。通过构建pGRB质粒,使其转录相应gRNA,从而与Cas9蛋白形成复合体,通过碱基配对与PAM识别目的基因,实现目的DNA双链断裂。
1.2.1 重组片段获得以及重组pGRB质粒构建
根据目的基因的序列,利用软件Primer Premier 5进行引物设计,通过PCR获得目的基因片段。以待整合或待敲除位点上下游序列为模板,设计合适的PCR引物,扩增500 bp左右的上、下游同源臂片段。对目的基因片段、上下游同源臂片段进行重叠PCR,以获得基因整合用的重组DNA片段。进行基因敲除时,只需对上下游同源臂片段进行重叠PCR构建。
使用含有靶序列的DNA双链片段与线性化的载体片段利用同源重组的方法构建pGRB重组质粒。使用CRISPR RGEN Tools网站设计靶序列,设计引物:5-线性化载体末端序列(15 bp)-酶切位点-靶序列(不包括PAM序列)-线性化载体末端序列(15 bp)-3’及其反向互补的引物,单链DNA通过退火获得包括目的序列的DNA双链片段。利用ClonExpress®II One Step Cloning Kit重组酶将靶序列与线性化的pGRB载体进行重组连接,通过化学转化的方法导入E.coliDH5α化转感受态,筛选阳性转化子,得到含目标靶序列的pGRB载体。
1.2.2 构建重组菌株
将pREDCas9质粒通过电转化导入感受态细胞中,平板培养并筛选阳性转化子。将阳性菌株接种到含LB培养基的摇管中培养12 h,再接种于2 XYT培养基(蛋白胨16 g/L,酵母粉10 g/L,NaCl 5 g/L,奇霉素100 mg/L)中培养,待OD600长到0.1~0.2时加入异丙基-β-D-硫代半乳糖苷,OD600长到0.3~0.4时收集菌体,用10%的甘油洗涤,制备电转化感受态[10]。将pGRB质粒以及重组片段同时电转化至感受态细胞中,在含氨苄青霉素抗性和奇霉素抗性的LB平板上,32 ℃培养。待长出单菌落后,通过菌落PCR筛选阳性转化子,再消除pGRB和pREDCas9质粒,获得目标菌株。
1.3 L-丝氨酸工程菌株发酵
1.3.1 摇瓶发酵
首先将菌种接种在固体斜面培养基(蛋白胨10 g/L,酵母粉5 g/L,NaCl 10 g/L,牛肉膏10 g/L,蔗糖1 g/L,琼脂粉20 g/L),置于37 ℃培养箱中培养12 h后转移至新的固体斜面培养基再培养12 h。用接种环刮取一环菌体,接种于装有30 mL种子培养基的500 mL圆底三角瓶中,用九层纱布封口,37 ℃、200 r/min培养10 h。种子培养基成分为:葡萄糖20 g/L,酵母粉2 g/L,蛋白胨2 g/L,KH2PO4·3H2O 1.2 g/L,MgSO4·7H2O 0.5 g/L,FeSO4·7H2O 10 mg/L,MnSO410 mg/L,维生素B11.3 mg/L,维生素B31.3 mg/L,维生素B51.3 mg/L,维生素B71.3 mg/L,维生素B91.3 mg/L,维生素H 1 mg/L,苯酚红8 mg/L,消泡剂1滴,pH维持在7.0~7.2。将培养好的种子培养液按照10%的接种量接种于30 mL发酵培养基,在37 ℃、200 r/min条件下培养至发酵结束。发酵培养基成分为:葡萄糖20 g/L,KH2PO4·3H2O 2 g/L,MgSO4·7H2O 2 g/L,FeSO4·7H2O 20 mg/L,MnSO420 mg/L,维生素B12 mg/L,维生素B32 mg/L,维生素B52 mg/L,维生素B72 mg/L,维生素B92 mg/L,维生素H 2 mg/L,苯酚红8 mg/L,消泡剂1滴。在发酵过程中,根据培养基中苯酚红的颜色指示,补加氨水,使发酵液pH值维持在7.0左右。每当葡萄糖耗尽后,补加1 mL 600 mg/mL葡萄糖溶液维持发酵进行,发酵周期为24 h。
1.3.2 发酵罐补料分批发酵
将菌株活化之后使用无菌水冲洗菌体,接种于发酵罐中,发酵罐的种子培养基与摇瓶种子培养基相同。培养过程中通过自动流加质量分数为25%氨水,将pH值稳定在7.0左右,温度恒定在37 ℃,溶氧在20%~30%。菌体OD600达到10~12时,按照20%的接种量接种至发酵培养基,发酵罐发酵培养基与摇瓶发酵培养基相同。整个发酵过程溶氧维持在20%~30%,pH值维持在7.0,温度维持在37 ℃,当罐中的葡萄糖耗尽时,通过流加质量浓度800 mg/mL的葡萄糖溶液,维持罐中葡萄糖的质量浓度在0.1~2 g/L内。
1.3.3 发酵过程中的检测与分析
利用分光光度计测量发酵液在600 nm处的吸光度(OD600),根据已有测试实验,将菌株OD600值乘以0.39换算为细胞干重(dry cell weight,DCW)值。发酵液中的残糖量使用生物传感器进行检测。采用Sykam S433D氨基酸分析仪检测发酵液中L-Ser浓度[11],具体检测按照厂商提供的标准操作流程进行。
1.3.4 发酵数据统计学分析
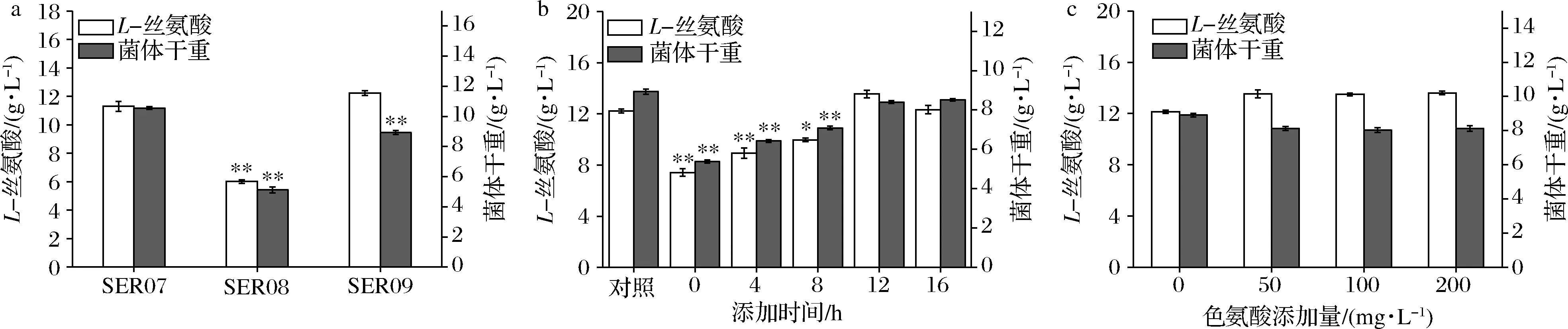
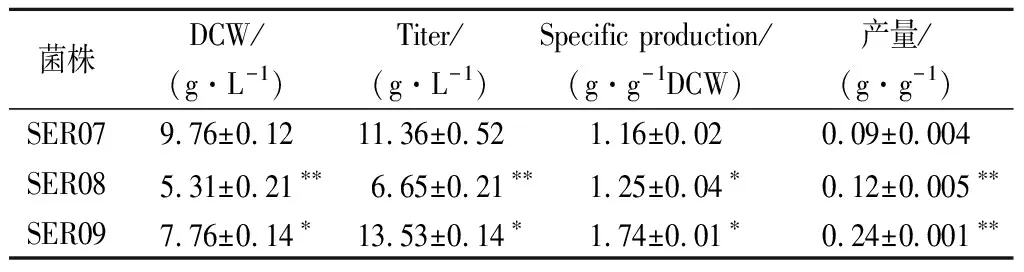
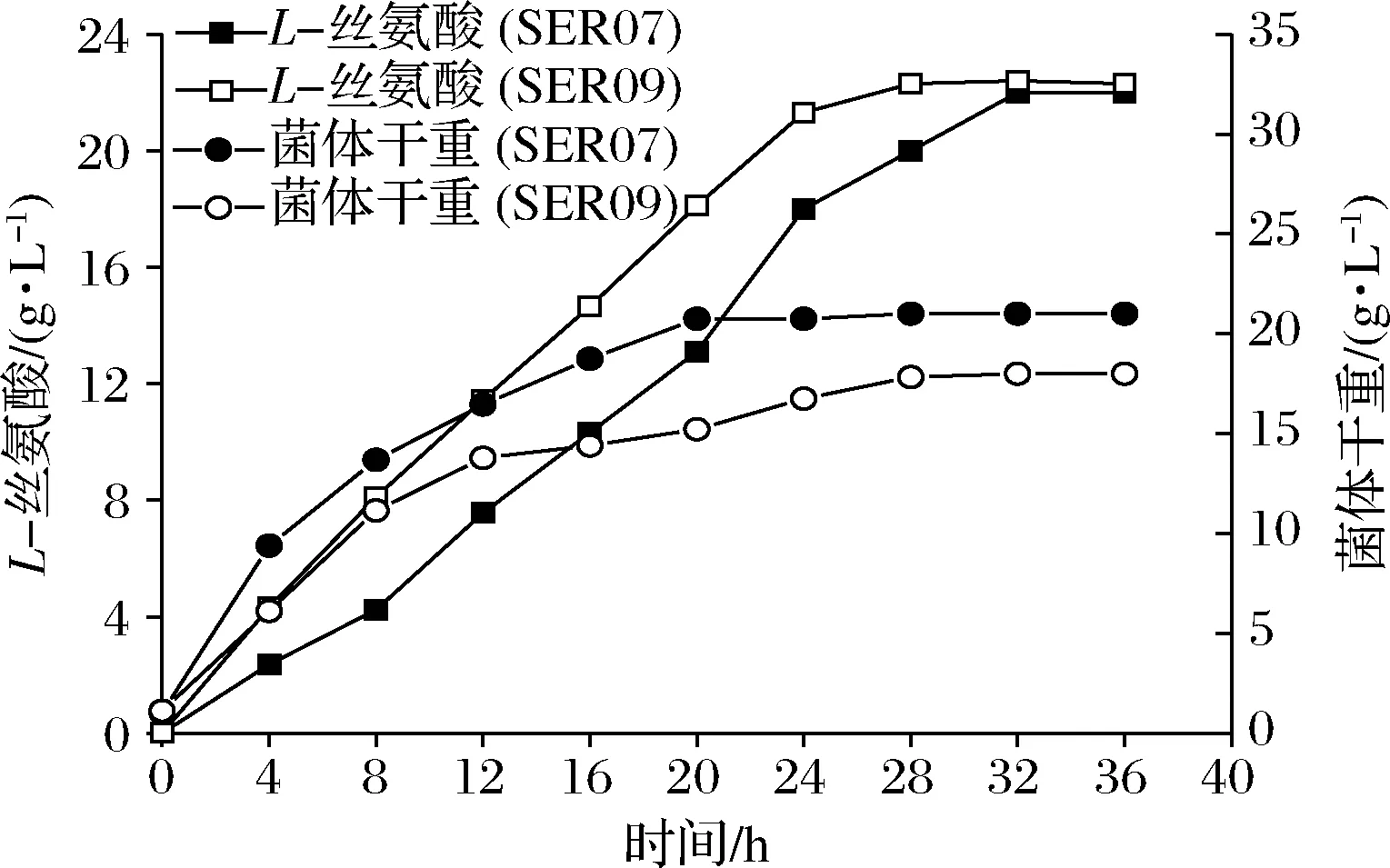
发酵数据代表3组平行发酵数据的平均值和标准偏差。利用t检验双尾分布对改造菌和对应出发菌的发酵结果进行单向方差分析。0.01 大肠杆菌中L-Ser可由sdaA,sdaB以及tdcG基因编码的丝氨酸脱氨酶降解为丙酮酸,也可由丝氨酸羟甲基转移酶(SHMT,由glyA基因编码)降解为甘氨酸[12-13]。本研究先将野生型大肠杆菌MG1655中编码丝氨酸脱氨酶的3个基因sdaA,sdaB以及tdcG进行敲除,阻断L-Ser向丙酮酸的降解,获得菌株SER01。摇瓶发酵结果显示(图2),发酵结束后,两株菌的细胞干重没有明显的差异,而3个基因敲除后,SER01可积累0.12 g/L的L-Ser,是出发菌株的2.77倍,说明阻断L-Ser向丙酮酸降解有效促进了L-Ser的积累,且对菌体的生长无影响。 图2 阻断L-Ser向丙酮酸的降解对L-Ser发酵的影响Fig.2 Effect of blocking the degradation of L-Ser to pyruvate on L-Ser fermentation 增加产物合成相关酶类的表达是构建工程菌种的常见策略。胞内3-磷酸甘油酸依次被磷酸甘油酸脱氢酶(PGDH,由serA基因编码),磷酸丝氨酸转氨酶(PAST,由serC基因编码)以及磷酸丝氨酸磷酸酶(PSP,由serB基因编码)催化生成L-Ser[14]。其中PGDH为L-Ser合成限速关键酶,其活性受到L-Ser的反馈抑制。根据文献报道[15],将PGDH中的第344位的组氨酸以及第364位的天冬酰胺替换为丙氨酸,可解除终产物对关键酶的反馈抑制。因此,本研究将该突变体的编码基因serA*整合到假基因位点yjiP,并由强启动子Ptrc启动转录,获得菌株SER02。摇瓶发酵结果显示,SER02的L-Ser产量为1.23 g/L,相比SER01提高了9.25倍;细胞干重为8.69 g/L,相比SER01降低了30.14%。结果说明serA*强化表达能增强L-Ser合成代谢,但对细胞生长存在一定程度的抑制作用。接着在SER02的假基因yjiV和ycjV位点分别整合了serC,serB基因,并由强启动子Ptrc启动转录,获得菌株SER03。摇瓶验证结果显示(图3),SER03的L-Ser产量达到5.41 g/L,是菌株SER02的4.39倍。 图3 强化L-Ser合成酶的表达对L-Ser发酵的影响Fig.3 Effect of enhancing the expression of L-Ser synthases on L-Ser fermentation 结果说明serC,serB基因表达的强化,显著增强了L-Ser的合成途径,有效促进了L-Ser的积累。另外,相比于对SER02,SER03的细胞干重有所增加,说明serC,serB基因表达使L-Ser的合成途径更为通畅,一定程度上缓解了关键基因serA*的单独表达所造成的代谢不平衡。为了进一步加强L-Ser的合成途径,本研究在假基因rph位点对serA*进行双拷贝,由强启动子Ptrc启动转录,获得菌株SER04。摇瓶验证结果显示(图3),SER04的L-Ser产量和细胞干重都大幅降低,说明serA*基因转录表达的强度过大,会使胞内代谢严重失衡,不利于菌体生长和产酸。 转运系统对氨基酸生产有重要影响,促进产物外排,减弱或阻断产物吸收途径是提高产物合成效率的重要举措[16]。高浓度L-Ser会对细胞产生生理毒性,及时将其从胞内转出有利于细胞正常生长。L-Ser与半胱氨酸结构相似,半胱氨酸转运蛋白EamA也可向胞外转运L-Ser[17]。本研究在菌株SER03的假基因tehB位点上,整合了eamA基因,由强启动子Ptrc启动转录,获得菌株SER05。摇瓶验证结果显示(图4),SER05的L-Ser产量达到7.41 g/L,较SER03提高了36.96%,说明强化转运蛋白EamA的表达,促进了L-Ser向胞外转运,有效促进了L-Ser的合成。另外,SstT和SdaC也是L-Ser的转运蛋白[18-19],可从胞外摄取L-Ser。于是本研究依次敲除了菌株SER05的sstT和sdaC基因,构建了菌株SER06和SER07。摇瓶验证结果显示(图4),SER06的L-Ser产量为8.65 g/L,相较于SER05提高了16.73%,SER07的L-Ser产量为11.23 g/L,相较于SER05提高了51.32%。结果表明sstT和sdaC基因的敲除显著促进了胞外L-Ser积累。 图4 转运系统改造对L-Ser发酵的影响Fig.4 The effect of transport system modifications on L-Ser fermentation L-Ser可由SHMT降解为甘氨酸,制约着L-Ser的大量积累。为了阻断该降解途径,促进L-Ser的合成,本研究首先尝试将SER07的glyA基因敲除,构建了菌株SER08。摇瓶发酵结果显示(图5-a),敲除glyA基因后菌体的生长和产酸都显著下降。这是因为L-Ser裂解为甘氨酸的反应是一碳循环的重要一环[20],对细胞代谢意义重大。因此选择合适的方式调控SHMT表达,平衡细胞生长与产物积累之间的关系,才会有效促进L-Ser的积累。 目前通过调节glyA基因表达来增加L-Ser积累已有较多报道,包括构建叶酸缺陷型[6]或者甘氨酸缺陷型[20]等方法,但是所得菌株在发酵过程中即便补加了适量的叶酸或者甘氨酸,菌体生长还是会受到一定的抑制,导致这些方法对L-Ser产量提升的效果有限。大肠杆菌色氨酸的合成受到严谨的多重调控,在转录水平上,色氨酸操纵子基因的转录会受到转录弱化和转录阻遏两种机制的调控。这两种调控机制都和色氨酸操纵子的启动子Ptrp相关,而Ptrp的强度受胞内色氨酸浓度的负调控[21]。为此本研究将glyA基因自身启动子替换为Ptrp,使glyA基因在菌株生长前期可以表达,保证细胞正常生长,待发酵至一定阶段后,通过向培养基中添加色氨酸弱化甚至阻断glyA基因的转录表达,减少L-Ser向甘氨酸的代谢,以期增加L-Ser积累。 将菌株SER08的glyA基因整合到假基因位点yeep上,用Ptrp启动子启动转录,获得菌株SER09。发酵结果显示(图5-a),菌株SER09的L-Ser产量为12.23 g/L,比SER07(原启动子控制glyA基因)提高了8.28%,DCW为8.93 g/L,比SER07降低了15.43%。因此总体来看,SER09的单位菌体产酸相比SER07提高了50%。更为明显的效果是,SER09的总耗糖为54.54 g/L,比SER07(109.0 g/L)降低了49.96%,因此糖酸转化率提高了1.15倍。随后本研究通过色氨酸的添加进一步调控SHMT的表达。首先在0,4,8,12,16 h分别添加200 mg/L色氨酸,以不添加色氨酸作为对照组,考察了色氨酸添加时间对L-Ser发酵的影响,结果显示(图5-b),发酵液中L-Ser积累量随着色氨酸添加时间的延长而增加,在发酵12 h时添加色氨酸效果最好。过早添加色氨酸可能会导致SHMT活性太弱,严重影响菌体生长,导致L-Ser产量也相应降低;过晚添加色氨酸则无法有效调控SHMT的表达,于是最终选择在发酵12 h时添加色氨酸。接着对色氨酸的添加量进行测试,结果显示(图5-c),培养基中添加50 mg/L色氨酸即可满足实验需求。最终在12 h添加50 mg/L色氨酸,SER09的L-Ser产量达到13.53 g/L,相比SER07,糖酸转化率提高了140%,单位菌体产酸提高了50%(表2)。 a-SHMT不同调控方式对L-Ser发酵的影响;b-不同色氨酸添加时间对L-Ser发酵的影响;c-不同色氨酸添加量对L-Ser发酵的影响图5 启动子Ptrp调控SHMT表达对L-Ser发酵的影响Fig.5 The effect of SHMT expression regulated by Ptrp promoter on L-Ser fermentation. 表2 启动子Ptrp调控SHMT表达对L-Ser发酵的影响Table 2 The effect of SHMT expression regulated by Ptrp promoter on L-Ser fermentation 为测试菌株的综合性能,将菌株SER07和SER09进行5 L发酵罐发酵测试,在发酵12 h时添加50 mg/L色氨酸。发酵结果显示,SER09虽然菌体生长速度变慢,但是最终的DCW值和SER07差距不大,并且在发酵前期SER09产酸速率明显高于SER07,28 h时SER09产酸达到最高值为22.31 g/L,SER07在32 h时产量达到最高值22.07 g/L(图6)。28 h时SER09的糖酸转化率为0.19 g/(g葡萄糖),比菌株SER07(32 h)提高84.38%,发酵结束时SER09的单位菌体产酸为1.25 g/g DCW,比菌株SER07提高了25.19%。本研究利用Ptrp启动子启动glyA基因的转录表达,通过控制色氨酸的添加量和添加时间动态调控SHMT的表达,结果表明这种调控方式不会造成营养缺陷,可在满足菌体正常生长的前提下,有效降低SHMT的活性,显著增强了L-Ser的合成途径。另外,SER07和SER09在发酵后期L-Ser浓度达到20 g/L左右以后,L-Ser产量都不再增加,这可能跟胞内高浓度L-Ser会对细胞产生生理毒性有关。增强菌株对高浓度L-Ser的耐受性是后续菌种性能优化的重点。适应性进化可有效提高菌株的耐受性,其近年也被成功地应用于激活细胞潜在通路或者提高目标产物的产量[22]。YONGZE等[23]通过进化提高大肠杆菌KC01对乙醇耐受性,使所得突变体SZ407与对照菌株相比,细胞干重提高了67%,单位体积乙醇生产速率提高48%,最终产量提高94%,改造效果明显。适应性进化的方法也用提高菌株对高浓度L-Ser耐受的菌株[7]。因此为提高工程菌株SER09L-Ser的产量,可对其进行传代培养,在培养过程中逐步增加培养基中L-Ser的添加量,获得高耐L-Ser的菌株。 图6 工程菌SER07和SER09在5 L发酵罐上分批 补料发酵结果Fig.6 Fed-batch fermentation results of the engineered strain SER07 and SER09 in a 5 L bioreactor. 本研究对野生型大肠杆菌MG1655进行了系统的代谢工程改造,从头构建了1株L-Ser生产菌。主要是在基因组水平上,阻断了L-Ser向丙酮酸降解,增强了L-Ser合成酶类的表达并对L-Ser转运系统进行了改造。为进一步促进L-Ser的积累,本研究重点对SHMT的表达进行了调控:利用启动子Ptrp启动glyA基因的表达,在发酵12 h,菌体生长到一定程度后,通过外源添加50 mg/L的色氨酸,弱化glyA基因的转录表达。这种调控方式有效抑制了SHMT的活性,且未对菌体生长造成明显影响,因而显著提高了L-Ser的合成效率。工程菌株SER09是1株不含质粒,遗传背景清晰,无缺陷型的L-Ser生产菌株。最终所得工程菌株SER09在5 L罐上发酵28 h,可生产22.31 g/L的L-Ser,其糖酸转化率以及单位菌体产酸相较对照菌株SER07分别提高84.38%以及25.19%,且培养过程中无需额外添加甘氨酸,改造效果明显,适合作为平台菌株用于进一步的性能改良,具有较好的应用前景。另外,胞内高浓度L-Ser会对细胞产生生理毒性,这可能是发酵中后期L-Ser的质量浓度达到20 g/L左后停止增长的原因,因此在后期研究中计划利用适应性进化方法增强细胞的耐受性,利用高浓度L-Ser作为选择压力对工程菌株SER09进行传代培养,筛选具有一定耐受性的菌株,进一步提升菌株L-Ser的发酵性能。2 结果与分析
2.1 阻断L-Ser向丙酮酸的降解途径

2.2 强化L-Ser的合成酶类的表达

2.3 L-Ser转运系统的改造

2.4 SHMT表达的调控
2.5 工程菌发酵罐发酵测试
3 结论
