蒺藜苜蓿(Medicago truncatula)全长转录组测序及 分析
2021-09-14尚骁尧周玲芳尹芊芊晁跃辉
尚骁尧 周玲芳 尹芊芊 晁跃辉
(北京林业大学草业与草原学院,北京 100083)
蒺藜苜蓿是一种非常重要的豆科模式植物,其具有基因组小,自花授粉,生命周期短,结实率高,具有稳定的遗传转化体系,越来越受到科研工作者的青睐。蒺藜苜蓿有2个品种已经被测序成功,其中,A17最早于2011年测序完成,目前已经更新至4.0版本[1-2];另一个品种为R108,与A17相比,具有更高的遗传转化率,更短的生命周期,种子更易萌发,而且目前世界上最大的蒺藜苜蓿突变体库就是使用的R108,所以被广泛应用于基因功能研究,基于以上优点,R108蒺藜苜蓿于2014年被测序成功,并于2017年12月进行了版本更新[3-4]。
二代高通量测序(next-generation high-throughput sequencing,NGS)技术的出现,使高通量测序的成本有了大幅度的降低,使得转录组分析变得更加便捷。通过该技术,在蒺藜苜蓿中已经开展了大量的工作。通过NGS技术,分析一个MtTdp1α缺失的蒺藜苜蓿突变体,结果显示该突变体中与DNA修复,DNA损伤以及染色体重组相关的基因转录水平发生了显著的变化[5]。通过NGS技术分析蒺藜苜蓿根瘤的发生过程,鉴定出1 140个差异表达基因,其中很多涉及磷转移的基因表达水平明显上调;大量涉及根瘤形成的基因表达水平显著上调,而抑制根瘤形成的基因表达水平下调[6]。利用该技术,在蒺藜苜蓿长期添加细胞分裂素及细胞分裂素抑制剂的过程中,鉴定出大量与蒺藜苜蓿响应细胞分裂素相关的基因,这些基因为研究蒺藜苜蓿生长发育和激素调节提供了候选[7]。
研究表明,植物在转录水平具有更加复杂的过程,如转录前水平调控,主要包括一些可变性剪接(alternative splice,AS)和可变多聚腺苷酸化(alternative polyadenylation,APA)[8-9]。比 如 在拟南芥中,80%的含有内含子的基因存在可变性剪接[10],这些可变性剪接受到多种胁迫条件或者激素水平的调节[11-14],同样也发挥着重要的生物学功能。而这些内容难以通过NGS技术实现,因为NGS技术不能提供全长序列[15-16]。SMRT的出现,弥补了NGS技术的不足,因为通过SMRT可以获得基因全长mRNA序列,准确地区分不同的剪接亚型(splice isoform)及鉴定APA位点等。SMRT在很多物种中有了成功的应用,如通过该技术,进行了人转录组的深度分析,发现了大概14 000个可变剪接基因,其中有>10%是从来没有注释的[17]。应用该技术,在小麦中发现了3 026个新基因,在玉米中鉴定大量的新的基因亚型,大大丰富了这些植物基因组信息[18-19]。在一些豆科植物中也应用该技术进行全长转录组分析,如在红三叶中鉴定出5 492条可变性剪接,4 333条lncRNA以及3 762条融合转录本,这些信息在以往的基因组测序和二代转录组分析中从未出现[20]。虽然蒺藜苜蓿的基因组测序工作已经完成,但是AS、lncRNA、APA位点以及融合转录本信息的不足,如目前在蒺藜苜蓿R108中无融合转录本数据,以上情况严重制约了蒺藜苜蓿基因组深入发掘。
本研究利用SMRT技术对蒺藜苜蓿进行全长转录组测序及分析,通过与蒺藜苜蓿参考基因组进行数据比对,鉴定新基因及转录本,分析蒺藜苜蓿中的AS类型并统计不同类型的数量,同时预测蒺藜苜蓿中lncRNA及融合转录本。这些转录组数据能够有效提高蒺藜苜蓿基因组注释水平,极大丰富基因序列及结构信息,旨为深入发掘蒺藜苜蓿资源提供有用资源。
1 材料与方法
1.1 材料
试验中使用的蒺藜苜蓿R108种子为Samuel Roberts Noble Foundation惠赠,使用草炭灰∶蛭石∶珍珠岩(1∶1∶1)作为营养土,植物材料放置于人工气候箱中,生长条件为:白天25℃ 16 h,晚上22℃ 8 h;相对湿度为50%-70%。为了获得更多的转录组测序信息,选取不同的植物组织(包括根、茎、叶、花、荚果);对蒺藜苜蓿进行不同的胁迫处理(100 mmol/L NaCl和10% PEG2000,处理1 d);对蒺藜苜蓿进行不同的激素诱导(10 μmol/L ABA、10 μmol/L GA3、10 μmol/L IAA和10 μmol/L 6-BA,处理6 h);分别收集不同的植物材料,混合为一个样品,进行全长转录组测序。
1.2 方法
1.2.1 建库及SMRT测序 通过植物RNA提取试剂盒分离植物样品总RNA,去除基因组DNA后,对提取的RNA进行纯度及完整性检测。吸取5 μg总RNA用于建库反应,基于大小分为2个文库(<4 kb与>4 kb),使用Pacific Bioscience Sequel平台对这2个文库进行SMRT测序。
1.2.2 数据处理 测序获得的原始数据(raw read)通过SMRTlink软件(version 4.0)进行处理,运行参 数 为:minLength=200,minReadScore=0.75。处理后的数据使用subread BAM files文件处理后,生成环状一致性序列(circular consensus sequence,CCS),参 数 设 置 为:parameters:min_length 200,max_drop_fraction 0.8,no_polish TRUE,min_zscore -999,min_passes 1,min_predicted_accuracy 0.8,max_length 18000。通过分析5'接头、3'接头及ploy(A)结构,鉴定出FLNC序列,经聚类、校正后,最终获得校正序列。
1.2.3 基因组比对及序列结构分析 SMRT测序结果,通过GMAP软件(version:2017-01-14)与蒺藜苜蓿R108参考基因组(R108_HM340_v1.0)进行比对。通过TAPIS pipeline软件(Version 1.2.1)进行基因结构及APA分析[21],通过SUPPAA软件(Version:2017-02-07)进行AS分析[22],通过MEME对转录本的poly(A)位点上游50碱基序列进行poly(A)信号分析,同时对转录本进行基因组跨区域分析,以鉴定融合转录本[19]。
1.2.4 开放阅读框、转录因子及lncRNA鉴定 校正后的序列的开放阅读框(open reading fragment,ORF)使用ANGEL pipeline进行分析,其中包含有完整ORF、5'非编码区及3'非编码区的转录本被鉴定为全长转录本,并进行编码氨基酸序列分 析[23]。对 转 录 组 数 据 使 用CPC[24]、CNCI[25]、PLEK[26]以及Pfam[27]4种方法进行lncRNA序列预测,并对预测的lncRNA进行分类[28]。下载已知的蒺藜苜蓿lncRNA(https://greenc.sciencedesigners.com/api/db/greenc/species/Medicago_truncatula/fasta),使用BLASTN程序进行序列比对以寻找新lncRNA,参数设置:e-value ≤1e-10,min-identity=90%,mincoverage=85%。
1.2.5 RT-PCR验证 为了验证SMRT分析的新基因及融合转录本预测结果,通过RT-PCR方法进行验证。提取蒺藜苜蓿总RNA,去除基因组DNA后,反转录为cDNA。基于SMRT分析结果,通过DNA Man(Version 6.0) 和Primer Premier(Version 5.0)软件进行引物设计。以cDNA为模板,进行PCR反应,待反应结束,取5 μL PCR产物经1%琼脂糖凝胶电泳分析,选取含有目标大小的PCR产物,送北京睿博生物技术公司测序。
2 结果
2.1 SMRT测序数据统计
通过对2个文库进行SMRT测序,一共获得了7 728 183个subread(共计11.77 Gb数据量),通过分析得知平均长度为1 524 bp,N50为2 051 bp (表1)。为了获得更精确的数据信息,这些数据生成了659 220条CCS,共鉴定出513 951条全长序列,其中509 014条为FLNC序列,平均长度为2 047 bp(表1)。这些FLNC序列被聚类为243 832一致性序列(ICE consensus,表1),联合非全长序列,共获得了243 676高质量的全长优化序列(polished consensus),平均长度为2 321 bp,N50为3 380 bp(表1);这些优化后序列经NGS序列校正后,共生成了236 243条校正序列(corrected consensus),这些序列平均长度为2 425 bp,N50为3 496 bp;对校正后的序列进行分析得知:其中220 233条(占93.22%)长度>500 bp,180 874条(占76.56%)长度>1 kb(表1)。
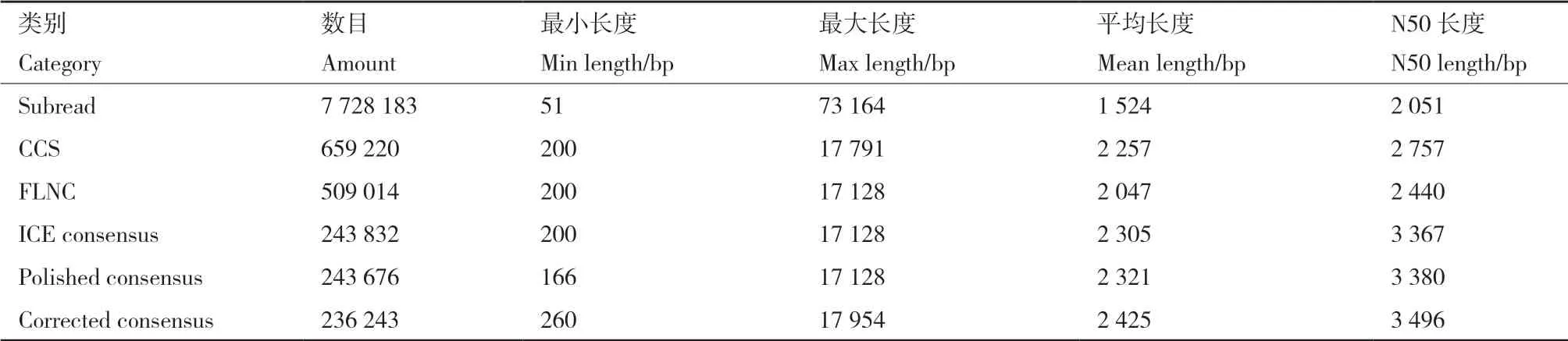
表1 SMRT测序数据统计Table 1 Statistics of sequencing data by SMRT
2.2 新基因及转录本鉴定
校正后的序列与蒺藜苜蓿基因组比对后,发现这些校正后的序列可以分为4类(图1):(1)未比对到参考基因组上的(unmapped)13 327条(占5.64%);(2)与参考基因组多个区域比对上的(multiple mapped)18 393条(占7.79%);(3)与参考基因组正向序列比对上的(mapped to ‘+’)148 971条(占63.06%);(4)与参考基因组反向序列比对上的(mapped to ‘-’)55 552条(占23.51%)。
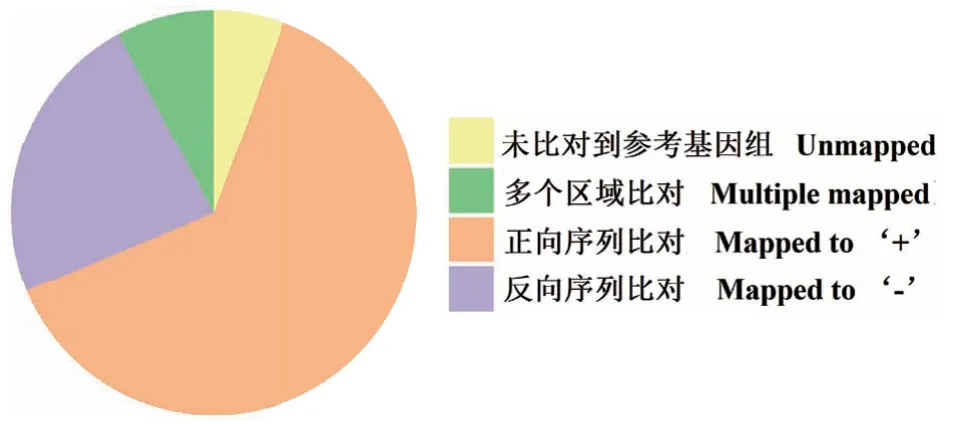
图1 校正后序列与参考基因组比对分析Fig. 1 GMAP analysis of corrected reads to reference genome
与参考基因组比对上的序列经过进一步的数据过滤(去除可能存在的错误序列及重复序列)后,共得到了61 125条最终序列(RC61125),这些序列可以分为3种类型(图2):(1)8 489条与参考基因组完全一致;(2)43 592条为已知基因的新转录本;(3)9 044条来源于新的基因。总之,通过SMRT测序共鉴定出7 209新基因以及52 636条新转录本。
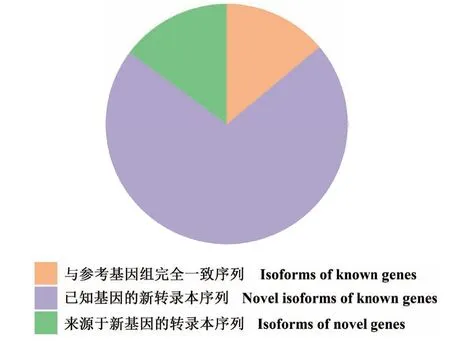
图2 RC61125序列的分类Fig. 2 Classification of RC61125 sequence
对SMRT测序鉴定的7 209新基因使用进行功能注释,结果显示,共有96.85%的新基因被注释成功(表2)。
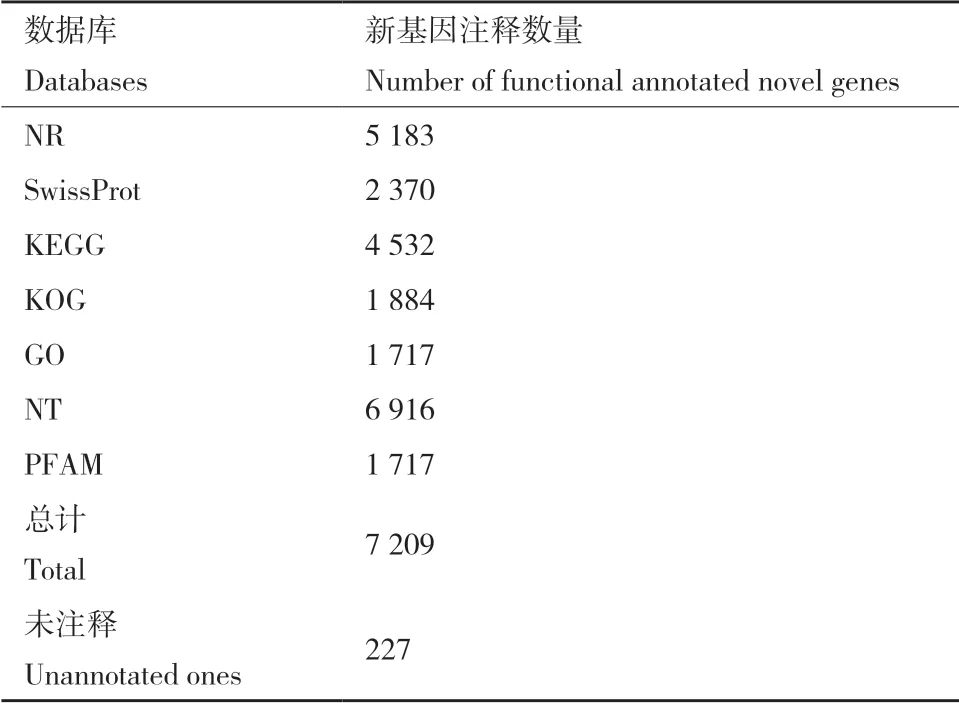
表2 新基因功能注释Table 2 Functional annotations of novel genes
将这些新基因与NR数据库比对,发现数量分布最多的前5物种为:蒺藜苜蓿A17(M. truncatula,4 128)、地三叶(Trifolium subterraneum,594)、木豆(Cajanus cajan,100)、鹰嘴豆(Cicer arietinum,70)和大豆(Glycine max,52),这些物种主要为豆科植物(图3)。KEGG分析显示:4 532个新基因可以富集到298条KEGG通路中;GO分析结果显示:1 717个新基因主要可以分为生物进程,分子功能和细胞组分。
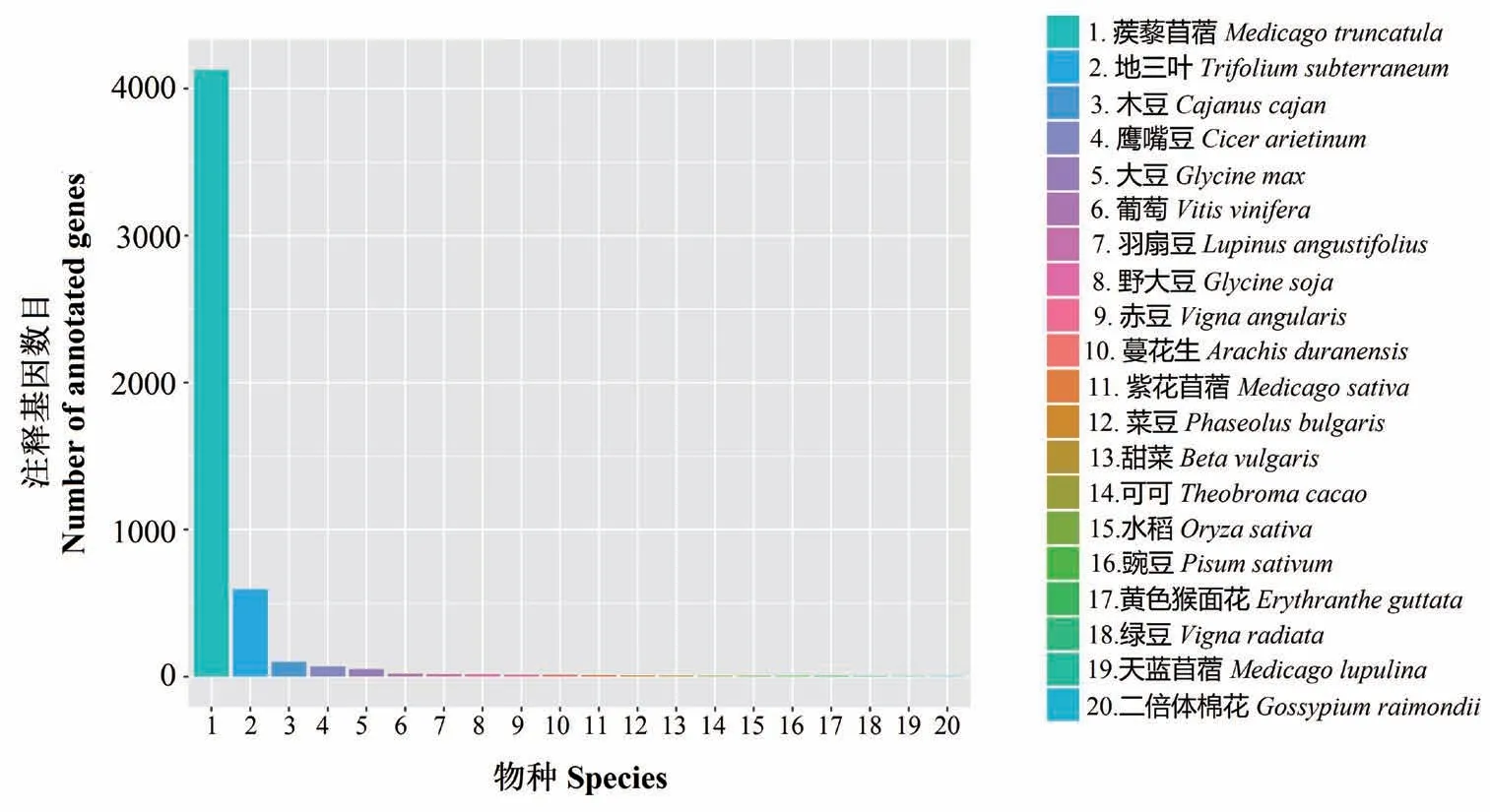
图3 新基因Nr同源物种分布图Fig. 3 Nr homologous species distribution diagram of novel genes
为了验证SMRT发现新基因的准确性,随机选择5个新基因进行RT-PCR验证。结果(图4)显示,基于SMRT技术发现的新基因序列,设计的引物,均能扩增出目标大小DNA片段,PCR产物经生物技术公司测序也验证了序列的正确性。以上结果表明,发掘蒺藜苜蓿新基因,SMRT是一种可靠的技术手段。
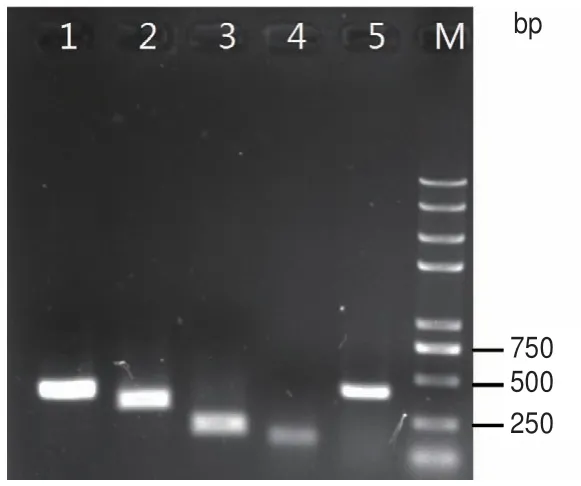
图4 新基因RT-PCR验证Fig. 4 RT-PCR validation of novel genes
2.3 基因结构分析
ORF分析显示共鉴定出36 235条编码蛋白质序列,这些序列平均长度为1 027.03 bp,其中13 451条含有完整编码区(表3)。对序列的5'-UTR及 3'-UTR数量与长度分布统计结果显示含有11 071个5'-UTR,平均长度为992.86 bp,含有1 990个3'-UTR,平均长度为799.00 bp(表3)。
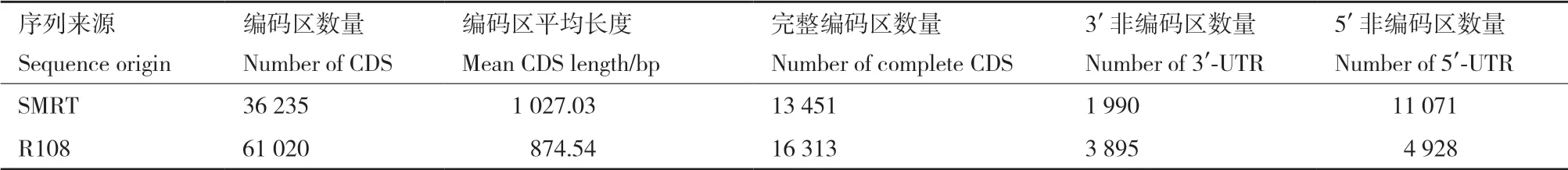
表3 编码区及非编码区鉴定Table 3 CDS and UTR identification
RC61125用于外显子结构分析,结果(图5)显示,17 590条(占28.78%)只含有1个外显子,8 327条(占13.62%)含有2个外显子,外显子数目>20的序列为1 586条(占2.59%);基于已经公布的2种蒺藜苜蓿参考基因组序列分析,在R108中,15 865条(占26.00%)转录本只含有1个外显子,而外显子数目>20的序列仅为612条(占1.00%),在A17中,13 594条(占23.17%)转录本只含有1个外显子,而外显子数目>20的序列仅为696条(占1.19%,图5)。内含子数目及结构分析,结果显示,参考基因组R108中内含子数目为184 641个,平均每个基因3.31个,而SMRT共测得269 289个内含子,平均每个基因11.26个。

图5 转录本外显子分布Fig.5 Distribution of exons in transcripts
2.4 AS及splice isoform分析
通过AS分析显示,SMRT测序共鉴定了11 761个AS,该数量在参考基因组中仅为1 468个;AS类型分析显示:SMRT技术鉴定的主要AS类型为RI,该特征与其他植物一致,而参考基因组中主要AS为外显子跳跃(skipped exon,SE)(图6);与参考基因组比较发现,SMRT测序鉴定出9 582个新AS。
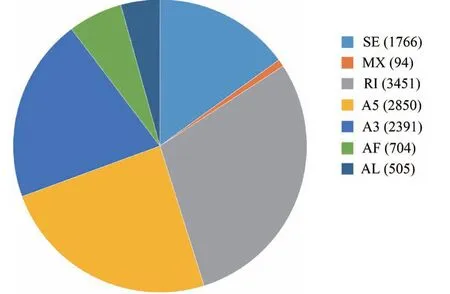
图6 AS数量及类型 Fig.6 Numbers and types of AS events
基因转录本分析显示,在参考基因组中,只有1条转录本的基因数目为52 332(占93.94%),而含有>10条转录本的基因数目为5,其中有2个基因(maker-Contig51-snap-gene-51.43 和maker-Contig137-exonerate_est2genome-gene-5.0)的转录本数目最多为13(表4);SMRT数据分析显示,含有1条转录本的基因数目为11 730(占49.03%),含有2条转录本的基因数目为4 713(占19.70%),而含有>10条转录本的基因数目为514(占2.15%,表4)。如基于SMRT分析发现,在蒺藜苜蓿 maker-Contig176-snap-gene-26.47基因中含有6条转录本,而在参考基因组中仅注释为1个。
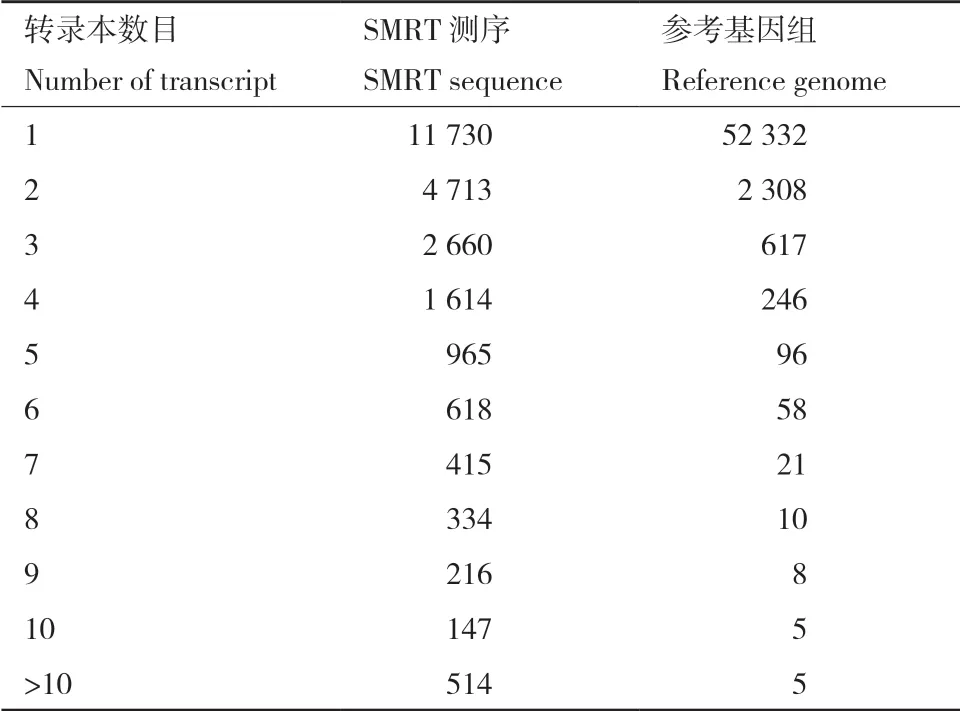
表4 SMRT测序及参考基因组转录本数目Table 4 Number of transcripts in SMRT sequence and reference genomes
2.5 lncRNA及融合转录本鉴定
通过4种方法,共鉴定出6 595条lncRNA,平均长度为1 250 bp,其中5 265条(占79.83%)仅含有1个外显子(图7-A)。根据基因组上的位置关系,将lncRNA分为4种类型(图7-B):(1)基因间区的lncRNA(intergenic lncRNA,lincRNA)3 669条(占55.65%);(2)反义lncRNA(antisense)779条(占11.8%);(3)内含子型lncRNA(intronic)340条(占5.16%);(4)与mRNA外显子有重叠区lncRNA(sense overlapping lncRNA)1 807条(占27.40%)。通过与参考基因组的注释lncRNA对比分析,发现共鉴定出6 503个新lncRNA。

图7 lncRNA鉴定Fig.7 Identification of lncRNA
融合转录本预测显示,通过SMRT分析共发现了479条融合转录本,而不同染色体间(443条)发生融合转录本的概率远远大于同一染色体内(36条)。为了验证SMRT发现融合转录本的准确性,随机选择了5个融合转录本,进行RT-PCR验证。结果(图8)显示,基于SMRT技术发现的融合转录本序列,设计的引物,均能扩增出目标大小的DNA片段,PCR产物经生物技术公司测序也验证了序列的正确性。
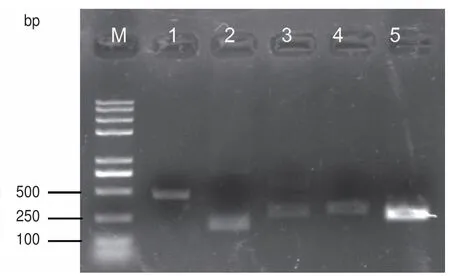
图8 融合转录本RT-PCR验证Fig. 8 RT-PCR validation of fusion transcripts
2.6 APA分析
通过分析发现,SMRT测序共鉴定了23 926个基因,其中12 049个基因(含有295 545转录本)至少存在1个poly(A)位点,441个基因存在至少5个poly(A)位点,平均每个基因含有2.14个poly(A)位点(图9)。对poly(A)剪切位点上、下游各50 bp核酸组成分析显示,蒺藜苜蓿poly(A)剪切位点上游富含尿嘧啶(uracil,U),下游富含腺嘌呤(adenine,A);通过MEME进行保守元件分析,结果显示,在蒺藜苜蓿poly(A)剪切位点上游存在2个保守元件(AAUAAA与UGUA,图9)。
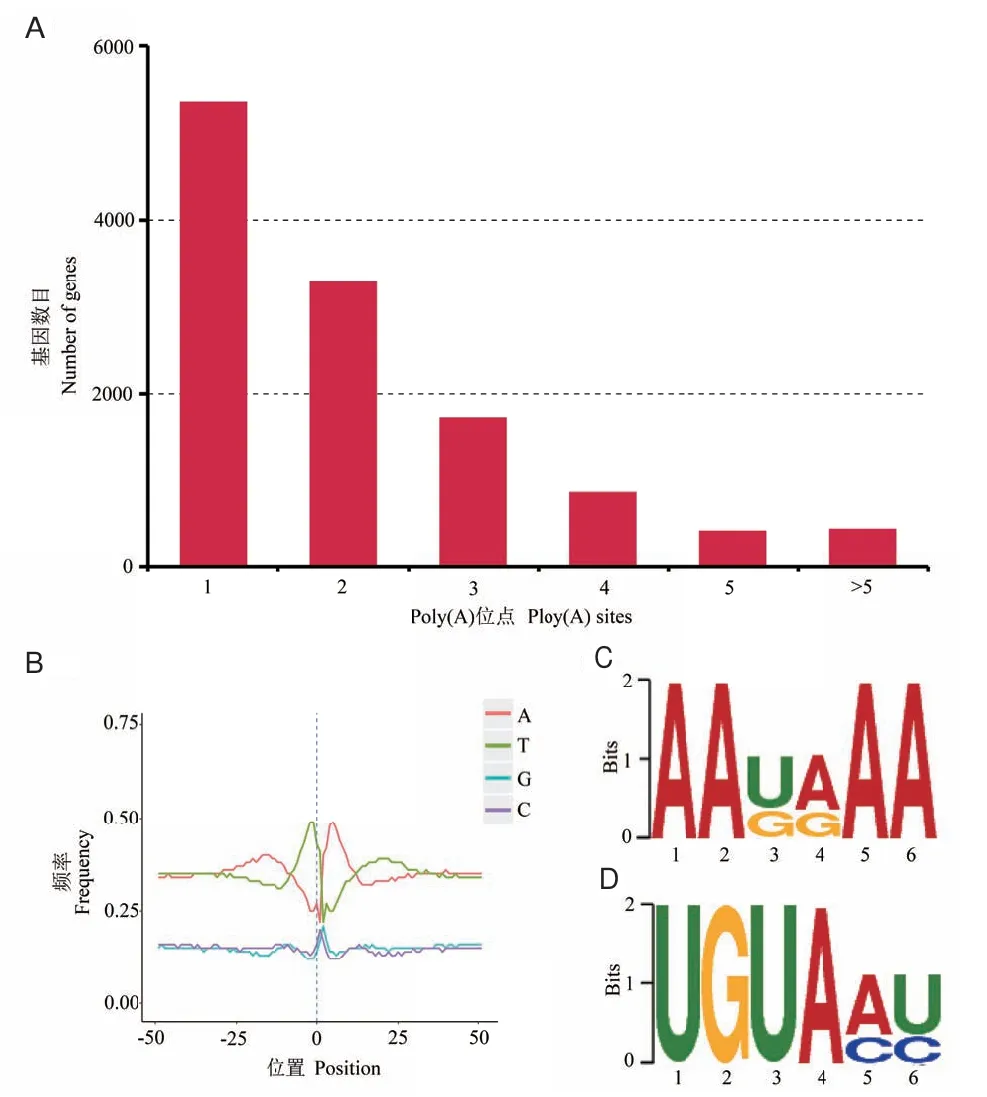
图9 poly(A)位点分析Fig. 9 Analysis of poly(A)sites
3 讨论
蒺藜苜蓿作为一种非常重要的豆科模式植物,虽然基因组序列已经公布,但是其转录组特征和mRNA结构并没有深入分析。随着测序技术的进步,SMRT为该部分工作的进行提供了新的途径。
全长转录组序列对于植物基因组注释和功能研究都具有极其重要的作用,通过SMRT对蒺藜苜蓿全长转录组进行测序,共产生了659 220 CCS,其中509 014被鉴定为FLNC转录本。FLNC序列的长度可以用于评估全长转录组建库的优劣,通过长度分析,发现FLNC的序列分布与2个测序文库长度大小一致(图1)。同时CCS与FLNC序列不但从长度上具有二代测序不可比拟的优点,同时可以避免二代测序中短序列拼接的带来的序列错误。通过分析蒺藜苜蓿二代测序数据,共获得了236 243条校正序列,其中76.56%长度>1 kb,13 451条序列含有完整ORF,该数字远远低于三代测序的质量。通过SMRT测序,共鉴定出23 926个基因,平均长度为4 307 bp,不但比二代测序长度要高,而且比参考基因组中还要高2 076 bp。通过SMRT对其他植物测序研究,同样也支持了类似的观点,如:使用SMRT对小麦转录组测序分析工作,结果显示,测序平均长度比小麦参考基因组注释序列要高45 bp[21]。对紫花苜蓿同时进行SMRT及二代测序和分析,其中75.68%的二代测序长度低于1 000 bp,而SMRT测序中仅有3.76%序列长度低于1 000 bp[29]。
SMRT测序在新基因与新转录本发现上也具有很大的优势。通过SMRT测序,在毛竹中发现了8 091个新转录本[30],在穿山甲中鉴定出8 014个新基因[31]。本研究中,鉴定出了新基因7 209个,已知基因的新转录本43 592个及新基因新转录本9 044个。RT-PCR及PCR产物测序,也进一步证实SMRT测序在新基因及新转录本鉴定上的准确性。SMRT测序还能更好的分析AS,使用该技术,在蒺藜苜蓿中共鉴定了11 761 AS,而在参考基因组中该数目仅为1 468。其他植物中公布的数据来看,主要的AS类型为RI,如玉米[19]、毛竹[30]、草莓[32]、无油樟[33]等,而蒺藜苜蓿R108参考基因组的数据显示,主要的AS类型为SE。通过SMRT测序得知,蒺藜苜蓿R108中AS中发现9 582个新AS,而RI出现频率最高。在蒺藜苜蓿R108中共鉴定了479条融合转录本,融合转录本发生在不同染色体间的频率明显高于染色体内。蒺藜苜蓿转录本的融合特征与在玉米中发现的规律一致[19]。lncRNA作为目前研究的热点,在植物生长发育中起着重要的调控作用[34-36],本研究共鉴定了6 595条lncRNA,主要类型为lincRNA。蒺藜苜蓿参考序列中已经注释的lncRNA为9 676个,经过比对分析,通过SMRT鉴定的lncRNA中92个为已知lncRNA,而6 503个为新lncRNA。SMRT鉴定的lncRNA平均长度为1 250 bp,最长为8 175 bp,而目前已注释的lncRNA平均长度为372 bp,最大长度为3 890 bp。该结果与SMRT技术在其他植物中的发现一致,在玉米中lncRNA能达到6 kb[19]。通过SMRT技术共鉴定了23 926个基因,其中12 049个基因能够产生295 545条转录本,这些基因至少含有一个poly(A)位点。这些数据丰富了蒺藜苜蓿转录组信息,同时也为蒺藜苜蓿基因组版本的升级提供了支持。
4 结论
通过SMRT技术对蒺藜苜蓿进行全长转录组测序及分析,共鉴定出7 209新基因,52 636条新转录本,9 582个新AS,6 503个新lncRNA及479条融合转录本;对基因结构和APA特征进行了分析;对蒺藜苜蓿R108参考基因组进行了数据补充,同时将原有的主要的AS类型修正为RI。
