紫外分光光度法测定皂布叶中总蒽醌含量
2021-09-14刘安韬刘喜华卢月妹
张 婷 刘安韬▲ 刘喜华 卢月妹
1.广西卫生职业技术学院药学系,广西南宁 530023;2.右江民族医学院,广西百色 533000
皂布叶为鼠李科植物尼泊尔鼠李[Rhamnus napelensis(Wall.)Laws.]的全株,主治风湿关节痛、慢性肝炎、肝硬化腹水等病症[1],刘安韬等[2]对皂布叶的研究表明,皂布叶有良好的抗痛风作用。皂布叶中主要成分为蒽醌类化合物[3],但目前未见皂布叶蒽醌类化合物的测定方法,本文采用醋酸镁-甲醇显色法,建立了超声提取-分光光度法测定皂布叶中总蒽醌含量的方法,为该药材的质量研究和产品开发提供科学依据。
1 仪器与材料
1.1 仪器与试剂
普析通用TU-1901紫外可见分光光度计(北京普析通用仪器有限公司),KQ2200E超声波清洗器(昆山市超声仪器有限公司),DFT-100高速粉碎机(大德药械有限公司),赛多利斯分析天平BSA8201[赛多利斯(上海)贸易有限公司],XS204电子天平[梅特勒-托利多国际贸易(上海)有限公司],HH-S2数显恒温水浴锅(金坛市医疗仪器厂)。
1,8-二羟基蒽醌(对照品,中国食品药品检定研究院,批号:110829-9702);醋酸镁、无水乙醇、甲醇、乙酸乙酯(均为分析纯),均购自成都市科隆化学品有限公司。
共收集了12批次皂布叶样品,采收于广西武鸣县,靖西县等地,分别编号为1~12号,干燥粉碎后备用。见表1。

表1 皂布叶样品信息表
1.2 试剂及溶液配制
蒽醌标准溶液(0.1037 mg/ml):精密称取10.37 mg 1,8-二羟基蒽醌,至100 ml量瓶,加甲醇适量溶解,用甲醇稀释至刻度。
醋酸镁-甲醇溶液(质量分数为0.5%):称取2.50 g醋酸镁溶于500 ml甲醇中,储于棕色瓶中保存备用。
1.3 样品溶液制备
精密称取0.200 g样品粉末于100 ml锥形瓶中,加入50 ml 50%甲醇,超声(常温)提取30 min,放冷,补足重量,过滤。精密量取取25 ml提取液于烧杯中水浴蒸干至约2 min,加入15 ml 3.0 mol/L盐酸,水浴水解30 min,取出冷却后,用乙酸乙酯萃取10 ml(5 ml×2),合并乙酸乙酯液蒸干,加甲醇溶解,转移至25 ml量瓶,加甲醇至刻度。精密量取1 ml至50 ml量瓶,加4 ml醋酸镁-甲醇显色,加甲醇至刻度。
1.4 标准曲线的绘制
分别准确吸取蒽醌标准溶液1.00、2.00、3.00、4.00、5.00 ml置于50 ml容量瓶中,分别加入醋酸镁-甲醇溶液4 ml,摇匀显色,加甲醇定容至刻度,以试剂空白为参比,在230 nm处测定吸光度(A)。以蒽醌质量浓度(C)为横坐标(µg/ml),吸光度(A)为纵坐标,绘制标准曲线并拟合回归方程,结果见图1。从图1可知,醋酸镁-甲醇法测定总蒽醌的回归方程为:A=0.78C+0.0419,R2=0.9962,在0.002074~0.01037 mg/ml范围内线性关系良好。
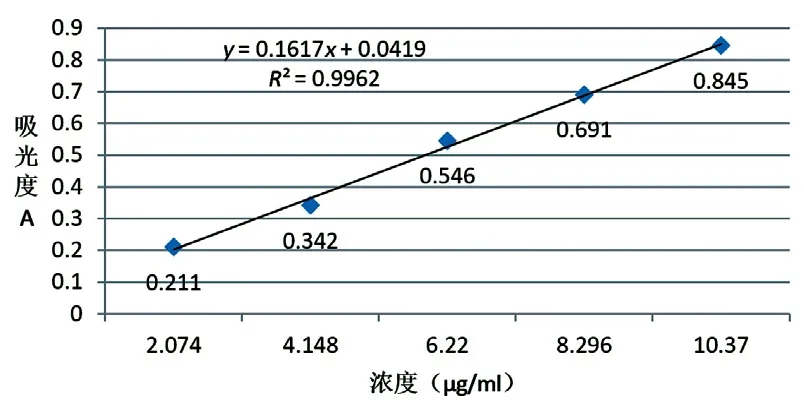
图1 标准曲线与回归方程
2 结果
2.1 测定波长的确定
吸取蒽醌标准溶液1 ml于比色管中,按照“1.4”项下显色后,于紫外可见分光光度计上扫描吸收曲线。可知,最大吸收波长为230 nm,因此,选择230 nm为测定波长,见图2。

图2 吸收光谱
2.2 提取溶剂对提取蒽醌浓度的影响
采用同一批次皂布叶样品Z4,按“1.3”项下超声提取法的步骤,考察了不同溶剂类型及溶剂浓度对检测结果的影响。分别选择甲醇和乙醇作为提取溶液剂,甲醇对皂布叶总蒽醌的提取效果明显好于乙醇;通过不同浓度甲醇提取浓度对比显示,50%甲醇提取的浓度更高,因此,选择超声提取的最佳溶剂和浓度为50%甲醇,见表2。
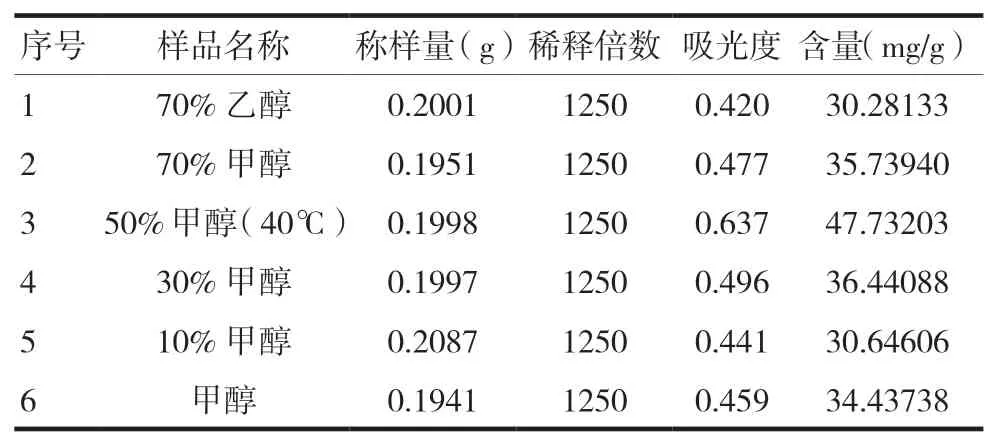
表2 不同溶剂类型及溶剂浓度对提取蒽醌质量浓度的影响
2.3 提取时间及温度对提取蒽醌浓度的影响
采用同一批次皂布叶样品Z4,按“1.3”项下超声提取法的步骤,考察了超声提取温度及时间对检测结果的影响。随着提取时间的增加,皂布叶中蒽醌浓度有明显的提高,但提取时间超过50 min后,蒽醌类氧化变色浓度开始明显下降;提取温度对于皂布叶中总蒽醌提取浓度影响不明显。因此,选择超声提取的最佳时间为30 min,选择超声提取的最佳温度为40℃,见图3及表3。
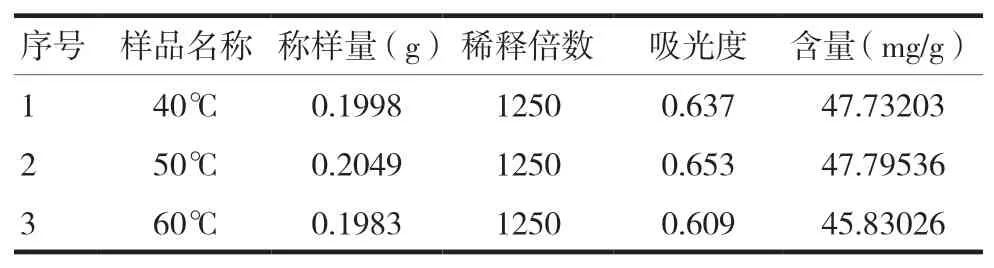
表3 提取温度对提取蒽醌浓度的影响

图3 超声提取时间对提取蒽醌浓度的影响
2.4 显色剂(醋酸镁溶液)用量对蒽醌浓度测定的影响
采用同一批次皂布叶样品Z7,取0.200 g按“1.3”项下超声提取法的步骤,考察了不同显色剂用量对检测结果的影响。不同显色剂用量对于皂布叶中蒽醌浓度测定有一定影响,在药材取样0.200 g时,用醋酸镁4 ml,测定结果稳定,效果良好。见表4。
2.5 方法学考察
2.5.1 稳定性 取标准曲线中6.22 µg/ml的对照品溶液,照“1.4”项下操作,在230nm波长下,分别在0、2、4、8、10、12、24、48 h测定其吸光度。吸光度RSD值为0.182%,见表5。

表5 稳定考察表
2.5.2 重复性 取2.5.1项0 h溶液,同法测定6次。结果吸光度RSD值为0.0747%。见表6。

表6 重复性考察表
2.5.3 加样回收率 精密称取皂布叶样品Z8,0.202 g,照“1.3”项下操作至精密量取1 ml,至50 ml量瓶,平行制备3份,分别加入0.002074、0.003111、0.004148 mg 1,8-二羟基蒽醌,加4 ml醋酸镁-甲醇显色,加甲醇至刻度,同法每份再平行配置两份。照“1.4”项下测定总蒽醌含量,计算加样回收率。平均回收率为95.71%,RSD值为1.24%,见表7。

表7 加样回收率考察表
2.6 样品含量测定
对12个批次皂布叶样品(1~12号)进行干燥粉碎后,按“1.3”项下操作,照紫外-可见分光光度法,在230 nm波长下测定,计算含量。皂布叶中总蒽醌的含量最高可达52.44 mg/g,最低为23.08 mg/g,平均含量为37.64 mg/g;对照药材采收表,皂布叶药材在9~10月份含量达到较高值,随着植物开花结果,全株药材的蒽醌物质含量有下降趋势,一般在1~2月降到最低,见表8。

表8 12个批次皂布叶样品总蒽醌含量
3 讨论
本研究建立了以超声提取-盐酸水解-乙酸乙酯萃取为预处理手段,以醋酸镁-甲醇显色测定皂布叶中总蒽醌质量浓度的方法,本方法操作安全、简单,操作效率高,且精密度好,结果稳定可靠。其中超声提取法广泛用于中药材中总蒽醌的提取[4-6],通过对影响提取率的温度、时间[7]、超声功率[8]和溶剂种类[9-10]、浓度等因素的考察,采用单因素法、正交实验法或响应面分析法[11-12]优化实验方案,因此本研究在进行提取方法考查中仅对超声提取的温度、时间和提取溶剂等主要条件进行考查,并得到了良好的效果。
醋酸镁-甲醇法主要针对游离态蒽醌进行显色测定,提取液中总蒽醌与醋酸镁的充分结合会明显影响测定结果,因此本研究考查了醋酸镁溶液的用量,保证显色的稳定性,另外溶液中酸的存在也会影响降低吸光度值[13],因为酸会对蒽醌与镁离子生成的络合物稳定性产生影响,所以收集到的乙酸乙酯萃取液一定要用水洗到中性。
皂布叶中化学成分以多酚类为主,其中的蒽醌类成分显示出良好的抗炎,抑菌和抗氧化等[14-17]药理活性,本研究以前期皂布叶提取物抗痛风作用[2]为基础,针对药材中主要的有效成分蒽醌类化合物建立测定方法研究,皂布叶药材在9~10月份含量达到较高值,随着植物开花结果,全株药材的蒽醌物质含量有下降趋势,为进一步规范药材使用,规范药材质量有一定的现实意义。
采用紫外-可见分光光度法进行含量测定具有较好的专属性和广泛的应用范围,在确定吸收波长条件下可以实现快速侦测。但由于光谱法本身的条件限制,测定样品的浓度过高会影响测定的准确度,另外会有同吸收峰的物质所干扰,影响准确度。高效液相色谱(HPLC)主要利用物质理化性质上的差异进行分离和定量测定。HPLC专属性强,准确度更高,但在含量测定中,对样品的组分的结构、性质的明确性要求较高,测定成分更明确,所需时间长。本研究主要针对皂布叶药材中蒽醌类成分进行测定,而不是单体成分,测定方法便捷,结果稳定可靠,可以作为皂布叶药材质量控制的方法。在以后的研究中,课题组将进一步深入研究该药材的药效成分,建立专属性更强的测定方法。
