BSCL2突变致先天性全身性脂肪营养不良伴严重中枢神经系统受累1例
2021-09-11马明圣邱正庆
丁 娟,马明圣,邱正庆
(中国医学科学院 北京协和医学院 北京协和医院儿科, 北京 100730)
先天性全身性脂肪营养不良(congenital gener-alized lipodystrophy,CGL)又称Berardinelli-Seip 综合征, 1958年首次报道[1]。目前共发现4种基因突变可导致本病:AGPAT2、BSCL2、CAV1、PTRF, 分别引起4种临床亚型(CGL1~CGL4)。其中CGL2为常染色体隐性遗传,由位于11q13的BSCL2基因致病性突变引起[2]。该基因编码的seipin蛋白主要表达于脂肪组织,对脂肪细胞的分化、脂滴形成及维持正常形态起重要作用。此外seipin蛋白还在神经元细胞内表达,对维持神经元细胞的正常功能起重要作用。CGL2患者主要表现为全身脂肪组织显著减少,易继发糖代谢和脂代谢异常,部分患者可合并神经系统受累,表现为智力迟滞,少数患者可表现为癫痫、神经系统退行性变[3]。本文总结了1例伴神经系统严重受累的CGL2患者的病例资料,探讨其基因突变特点,以提高对该病的认识。
1 材料与方法
1.1 对象
研究对象为北京协和医院儿科(2017-08-01)收治的1例以全身脂肪营养不良为主要表现的患儿。根据患儿的病史及体格检查结果,符合CGL诊断。经患儿父母知情同意,抽取外周血,提取基因组DNA进行第二代基因测序分析及Sanger测序验证。本研究经北京协和医院伦理委员会批准(伦理审批文号:S-K1705)。
1.2 方法
1.2.1 实验室检查和影像学检查:由北京协和医院检验科和放射科完成,包括血常规、生化、胰岛素水平、腹部超声、心脏超声、骨龄、脑电图等。
1.2.2 第二代基因测序及Sanger测序分析验证:由范德瑞尔(北京)医学科技有限公司完成。提取患儿及其父母外周血基因组DNA,构建基因组文库,然后通过探针杂交捕获相关的目的基因(包括AGPAT2、BSCL2、CAV1和PTFF等CGL相关基因)外显子及相邻内含子部分区域,并进行富集。富集的目的基因片段通过下代高通量测序仪Illumina HiSeq进行测序,对与临床主诉相关的明确的致病性变异,采用Sanger测序对受检者及父母进行验证。
2 结果
2.1 临床资料
患者男,5岁8个月。异卵双胎之一,足月剖宫产,出生质量2.7 kg。母孕期平顺。患儿出生后即发现皮下脂肪菲薄、消瘦。渐出现肌肉显现。4月龄出现全身皮肤色素沉着,毛发增多、增粗。5月龄查体发现肝大,当地医院查肝功能异常,腹部超声提示肝大,头颅核磁共振未见异常,血串联质谱未见异常,染色体核型分析未见异常。1岁之后身高、体质量增长加速,智力及语言迟滞,16个月会走,2岁叫“爸爸”、“妈妈”,听不懂简单的指令,多动、易激惹,但仍在逐渐进步。4岁之后出现癫痫发作,见于清醒期,发作形式有:1)发作性反应减低,双眼茫然、表情呆滞、持续数秒至数十秒后突然恢复。发作后无意识障碍。2)上肢下垂一下,或点头一下,或四肢节律性抖动几下,每日发作数十次,发作后无意识丧失。5岁开始出现走路不稳、容易摔跤、持物手抖、吐字不清、认知功能倒退。
父母体健,非近亲婚配。异卵双胎弟弟发育正常,无皮下脂肪减少表现。家族中无类似疾病患者。
2.2 体格检查
体质量32 kg,身高140 cm,均大于同龄同性别儿第97百分位[4],头围54 cm。神清,全身皮肤偏黑,颈部、腋下、肘窝、腹股沟、腘窝等皮 肤皱褶处色素沉着,全身皮下脂肪消失,肌肉显现,背部及下肢多毛,脸颊及下颌消瘦,心、 肺查体未见异常,腹部膨隆,肝右肋下可扪及8 cm,质地中等,移动性浊音阴性。神经系统查体:神志清、多动。可与人交流、执行简单指令。吐字不清,可以说数个字的短句,不会计数。四肢肌力正常,下肢肌张力增高,步态不稳,指鼻试验不能配合,双侧膝腱反射对称引出,克氏征、巴氏征阴性。颈抵抗阴性。
2.3 辅助检查
血常规:白细胞为5.52×109个/L,中性粒细胞百分比为40.0%,淋巴细胞百分比为48.6%,血红蛋白139 g/L,血小板248×109/L。丙氨酸转移酶 (alanine aminotransferase, ALT) 207 U/L,天冬氨酸转移酶(aspartate aminotransferase, AST) 113 U/L,谷氨酰转肽酶(glutamyl transferase, GGT) 69 U/L,白蛋白(albumin, Alb) 52 g/L,三酰甘油(triacylglyceride, TAG) 1.82 mmol/L,总胆固醇(total cholesterol, TC) 4.73 mmol/L,高密度脂蛋白胆固醇 (high density lipoprotein cholesterol, HDL-C)1.21 mmol/L,低密度脂蛋白胆固醇(low density lipoprotein cholesterol, LDL-C)3.09 mmol/L ;肌酐(creatinine, Cr)21 μmol/L,血电解质正常;空腹血糖4.5 mmol/L,空腹胰岛素53.89 μlU/mL。腹部超声肝剑下8.3 cm,肋下8.3 cm,肝右叶斜径11.7 cm,肝实质回声均匀。脾厚4.7 cm,长径10.4 cm,肋下未及。心脏超声左心房增大。双肾超声未见异常。左腕X线骨龄相当于9岁。阴囊超声提示交通性鞘膜积液。因不配合头颅磁共振检查失败。
视频脑电图中枢(electroencephalogram,EEG)醒睡各期见大量旁中线区多灶性及广泛性2.5 Hz~3 Hz中-高波幅棘慢波、慢波散发或阵发,清醒期监测到频繁不典型失神发作、失张力发作、肌阵挛-失张力发作、肌阵挛及局部肌阵挛发作(图1)。
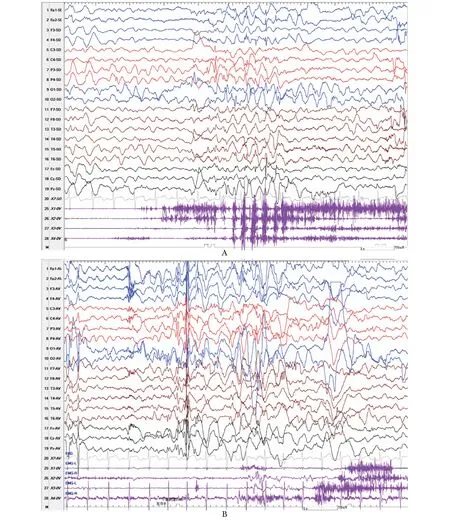
A.brief spike-slow waves discharges, and subcontinously myoclonic jerks on electromyogram(EMG);B.discharges of spike-waves associated with myoclonic jerks on the lower limbs of EMG
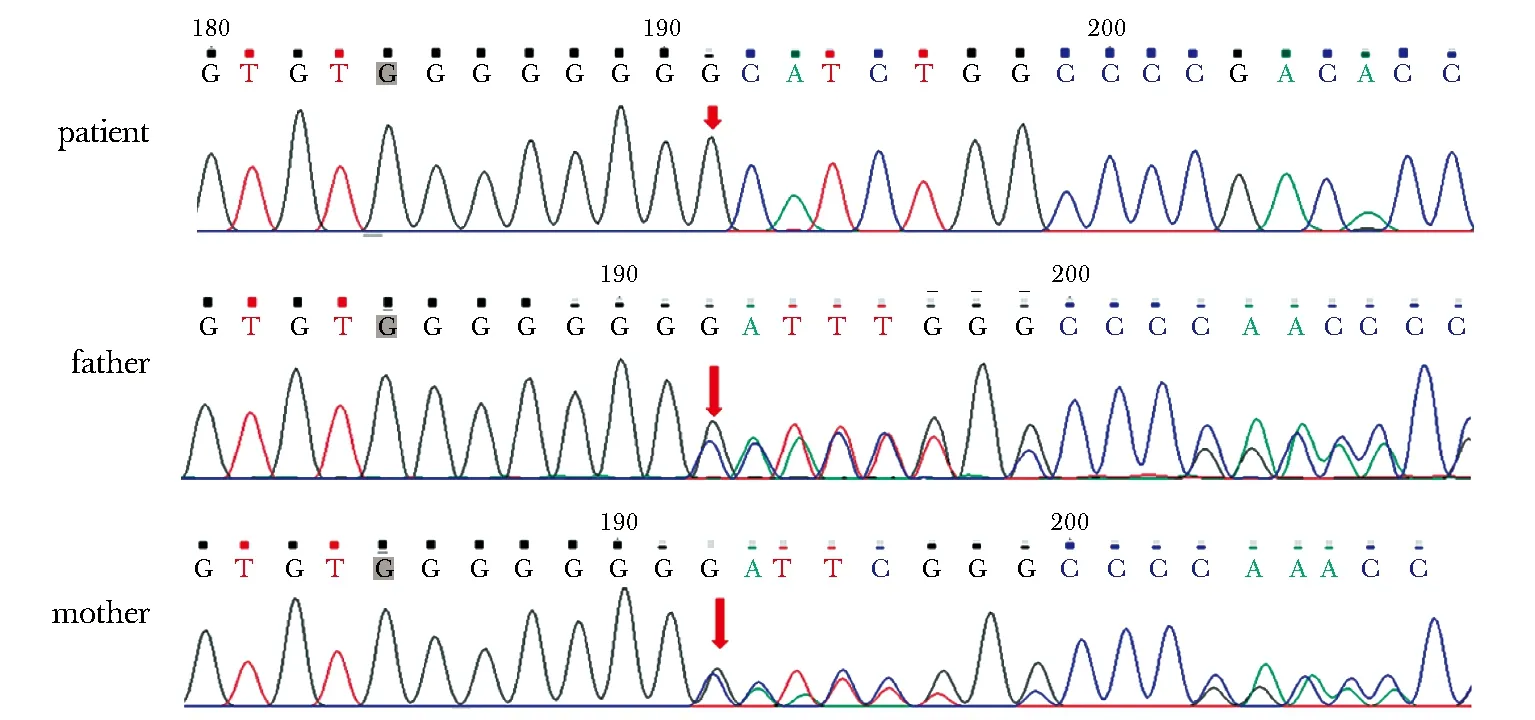
The red arrow showed that the homozygous frameshift mutations occurred in the BSCL2图2 患者BSCL2基因碱基重复纯合突变Fig 2 Patient’s homozygous base repeat mutations occurred in the BSCL2
2.4 基因检测
在该患者儿发现BSCL2基因c.782dupG碱基重复纯合突变(图2),父母该位点为杂合突变。此突变为移码突变,导致seipin蛋白第262位氨基酸由异亮氨酸变为组氨酸,从第262位氨基酸开始后面的第12位变为终止密码,是HGMD(human gene mutation database)报道的已知致病性突变。父母未发现除该位点以外的其他脂肪营养不良相关的基因突变。
2.5 治疗及随访
患儿诊断CGL2明确,给予低脂、低升糖指数饮食,口服二甲双胍,拉莫三嗪每日2次,每次75 mg 。半年后复查血生化:ALT 156 U/L,AST 98 U/L,GGT 77 U/L,Alb 50 g/L,TAG 3.88 mmol/L,TC 5.14 mmol/L, HDL-C 0.99 mmol/L,LDL-C 2.942 mmol/L,Cr 22 μmol/L;空腹血糖 5.1 mmol/L,空腹胰岛52.82 μlU/mL。癫痫发作仍频繁,发作形式同前,行走不稳症状较之前加重,认知功能倒退明显,生活不能自理,不能控制大小便。
3 讨论
本文报道了1例BSCL2基因 c.782dupG碱基重复纯合突变所致的CGL2疾患患儿,除典型的脂肪营养不良表现外,还合并严重神经系统受累,神经功能进行性恶化。
CGL2最常见的神经系统症状为智力迟滞,只有少数患者可合并难治性癫痫、严重脑病,最终在6~11岁死亡[5-9];其难治性癫痫符合进行性肌阵挛癫痫(progressive myoclonic epilepsy,PME)诊断,主要临床特点包括肌阵挛及多种类型的癫痫发作、进行性神经功能倒退。本患儿为中国首次报道的具有严重中枢神经系统伴癫痫发作的CGL2患儿,促进了对CGL2患者临床表型谱的认识。
CGL2患者脂肪缺失严重,临床表型重,很早即出现脂肪组织减少,肝大、肌肉肥大、心肌病、胰岛素抵抗和高TG血症是常见表现,患者可很快进展至糖尿病及肝硬化等。不同于其他类型CGL,CGL2常见智力发育迟滞,文献报道可达78%[3]。
目前文献报道CGL2合并PME患者的基因型主要分两种:1)c.782dupG纯合突变或复合杂合突变;2)c.985C>T纯合突变或复合杂合突变。Sánchez-Iglesias 等[7]对患者白细胞和成纤维细胞的分子研究发现c.782dupG碱基重复突变会导致外显子7跳跃,c.985C>T突变影响剪切位点,亦导致外显子7跳跃,由此推测外显子7跳跃可能是CGL2合并PME的致病机制之一[10]:外显子7跳跃导致短转录本表达增加,seipin蛋白正常寡聚作用受损,形成异常大分子聚合物,从而诱导神经元内质网应激(endoplasmic reticulum stress),产生神经损伤。已有新近研究报道其他基因型也可导致CGL2合并PME[11],由此可见致病机制并非仅与外显子7跳跃有关,CGL2的基因型与表型关系仍需更进一步研究。
根据典型的脂肪营养不良、BCL2基因检测出2个等位致病突变,可确诊CGL2。在获得基因检测结果前,需要与其他可引起脂肪消失的疾病相鉴别:1)获得性全身性脂肪营养不良,发生在先前健康的儿童或成人中,可见于HIV感染者或自身免疫性疾病患者。2)其他类型先天性全身性脂肪营养不良,脂肪营养不良严重程度及神经系统受累有所不同,基因检测可协助鉴别。
低脂等饮食控制对于该病的治疗非常重要。如有严重的高TAG血症可应用苯氧酸类药物,非高密度脂蛋白胆固醇水平升高可应用他汀类药物。合并糖尿病者可应用二甲双胍、磺脲类药物及胰岛素。重组人瘦素类似物已被国外批准用于治疗CGL2,有多个研究证实其可有效改善胰岛素敏感性,降低TAG,减少心、肝脂肪沉积,降低糖化血红蛋白及TAG水平。早期开始瘦素替代治疗可能有助于延缓脑病进展、改善神经系统预后[7,12]。多种抗癫痫药物如拉莫三嗪、丙戊酸钠、氯硝西泮、乙琥胺、苯巴比妥、吡仑帕奈等可用以控制发作,但是CGL2患者的癫痫通常难治,往往需要联用多种药物,迷走神经刺激术可能有部分效果[11]。阿立哌唑、苯海索、巴氯芬等可用来改善肌张力障碍[9]。本文报道的CGL2患儿虽然经过饮食控制、二甲双胍、拉莫三嗪等治疗,高TAG血症及胰岛素抵抗无改善,癫痫发作难以控制,认知及运动功能倒退明显。瘦素替代治疗可能改善该患儿状况,但该药在中国尚未上市。
综上所述,BSCL2等位突变可引起CGL2,该病除典型全身脂肪营养不良、代谢异常、智力迟滞表现外,还可合并PME、严重脑病。合并严重脑病者常见突变位点为c.782dupG和c.985C>T,但基因型与表型之间的关系仍需更多研究。
