心导管检查对小儿不同类型肺动脉高压的诊治价值分析△
2021-09-11李博宁欧振恒李薇玢
李博宁,刘 琮,谢 颖,欧振恒,辛 颖,李薇玢,姚 静
(深圳市儿童医院心血管内科,广东深圳 518038)
小儿肺高压是指3个月以上的小儿静息状态下在海平面平均肺动脉压力大于25 mmHg(1 mmHg=0.133 kPa)。小儿肺动脉高压则在小儿肺高压基础上另外要求肺毛细血管嵌压<15 mmHg,肺血管阻力指数<2 Wood·m2[1-3]。导致小儿肺动脉高压的原因包括先天性心脏病,基因遗传性、特发性结缔组织病,人类免疫缺陷病毒(HIV)感染,门脉高压以及药毒物等。心导管检查是诊断小儿肺动脉高压的“金标准”。目前,小儿肺动脉高压心导管检查报道较多[4-5],但对于不同类型肺动脉高压心导管患者的检查结果对比资料报道较少。本文总结深圳市儿童医院5 种不同肺动脉高压类型患者的临床资料,对比其心导管检查特点,以期对不同类型肺动脉高压的诊断及治疗进行临床指导。
1 患儿临床资料
患儿1,女,6 个月,5 kg,因“气促3 d”到深圳市儿童医院就诊,诊断为“先天性心脏病、室间隔缺损(膜周部,大小10 mm)、重度肺动脉高压、心功能III 级”,经过抗心力衰竭治疗,病情好转后,于2019 年11 月18 日行心导管检查术。患儿心导管检查术后第二天行外科手术治疗,手术顺利,患儿于2019 年12 月10 月痊愈出院。
患儿2,男,11 岁,24.3 kg,因“活动后气促3 年,发绀2 年”为主诉入住本院。诊断为“先天性心脏病、室间隔缺损(膜周部,大小15 mm)、重度肺动脉高压、心功能Ⅱ~Ⅲ级”。患儿于2019 年07 月29 日在本院行心导管检查术,心导管检查结果显示患儿肺动脉高压为非动力型,暂无手术指征,建议患儿口服波生坦治疗。目前患儿仍在随访中。
患儿3,男,9 岁,?kg,因“血尿1 个月余,发现肺动脉高压1 周”为主诉入住本院。患儿因血尿在外院治疗,肾脏穿刺病理诊断IgA 肾病,外院进行激素甲强龙冲击治疗时发现气促、浮肿,行心脏超声提示肺动脉高压,遂转入本院治疗。在本院行心脏超声检查提示肺动脉高压(重度,三尖瓣反流估测肺动脉压力为94 mmHg),给予波生坦+西地那非口服治疗,肺动脉压力降至40 mmHg 左右,再次行环磷酰胺静脉冲击治疗,监测肺动脉压力再次升高至90 mmHg以上。患儿心脏+肺部计算机断层扫描(computed tomography,CT)检查未见明显异常,排除解剖结构异常引起肺动脉高压。患儿C 反应蛋白、红细胞沉降率(ESR)、肾功能及自身免疫抗体均正常,人类免疫缺陷病毒阴性,排除因风湿免疫、血液肿瘤、人类免疫缺损病毒感染、代谢因素等引起肺动脉高压的可能。经多学科会诊讨论后,患儿于2019 年12 月13 日在本院心导管室行左、右心导管检查术。心导管检查结果显示左肺血管畸形导致肺动脉高压。追问患儿病史,患儿既往有习惯性鼻衄病史,肝脏中发现血管瘤,不排除遗传性毛细管扩张症可能,但基因外显子检查未见异常致病基因。患儿目前口服强的松,间断进行环磷酰胺冲击治疗IgA 肾病,持续口服波生坦+西地那非治疗,肺动脉压力波动于60 mmHg(超声监测)左右,无心功能不全症状。
患儿4,女,11 岁,46.5 kg,以“活动后气促伴胸闷1 年,加重5 d”为主诉于2019 年09 月10 日入住本院。心脏超声提示肺动脉高压(重度,三尖瓣反流估测肺动脉压力100 mmHg)。心脏CT 及肺部CT 未见心脏、双肺及血管异常。血液检查未见异常。血、尿代谢筛查未见异常。6 min 步行试验525 m(预期838 m)Borg 评分2 分,心功能评估Ⅱ级。2019 年09 月16 日行心导管检查术。基因检查提示骨形成蛋白Ⅱ型受体(bone morphogenetic protein type II receptor,BMPR2)变异。考虑BMPR2 相关性肺动脉高压。给予患儿联合波生坦+西地那非口服治疗,患儿经治疗胸闷好转,心功能好转后出院,目前仍在随访中。
患儿5,女,2 岁10 个月,10.2 kg,因“气促伴全身浮肿3 d”为主诉于2019 年06 月08 日入住本院。心脏超声提示肺动脉高压(重度,三尖瓣反流复测肺动脉压力94 mmHg)。心脏及肺部CT 未见心脏、肺部及血管异常。血液检查除血小板降低外,无其他异常。血、尿代谢产物筛查未见异常。给予静脉应用瑞莫杜林及输注血小板后患儿病情好转,后一直口服波生坦治疗。2019 年11 月20 日入院行心导管检查。基因检测提示活化素Ⅰ型受体(activator type I receptor,ACVR1)(c.789C>A,p.D263E)变异,考虑患儿ACVR1(c.789C>A,p.D263E)相关性肺动脉高压。目前口服波生坦治疗,2020 年1 月20 日(靶向药物治疗后7 个月)复查心脏超声提示肺动脉压力40 mmHg,患儿心功能不全症状。
患儿6,男,4 岁,16 kg,以“活动后气促1 年多,全身浮肿3 d”为主诉于2018 年05 月04 日入住本院治疗。心脏超声提示肺动脉高压(重度,三尖瓣反流估测肺动脉压力94 mmHg)。心脏CT 及肺部CT 未见心脏、双肺及血管异常。血液检查未见异常。血、尿代谢筛查未见异常。给予患儿波生坦+西地那非口服治疗,患儿心力衰竭好转后于2018 年05 月09 日行心导管检查。基因检测提示ACVR1(c.1450C>T,p.R484W)变异。考虑患儿为ACVR1(c.1450C>T,p.R484W)相关性肺炎。随访中,患儿单服波生坦+间断氧疗,肺动脉压力一度降为40 mmHg 左右。6 个月前,患儿曾患肺炎一次,肺炎期间肺动脉压力再次升至90 mmHg 左右,加服西地那非,并积极抗炎治疗,经治疗肺动脉压力逐渐下降。1 个月前患儿复诊,显示肺动脉压力为50 mmHg 左右,心功能尚可,目前继续为波生坦联合西地那非口服,间断氧疗,仍在密切随访中。
2 结果
2.1 不同类型患儿的心导管检查结果
患儿1 的PPA:PAO=0.9,肺毛细管嵌压(pulmonary artery wedge pressure,PAWP)=12 mmHg,肺动脉阻力指数(pulmonary vascular resistance index,PVRI)=3 Wood·m2,急性肺血管扩张试验(acute pulmonary vasodilation test,AVT)(+)。左右肺动脉分支手推造影剂行选择性肺小动脉造影(selective pulmonary arteriography,SPA)显示肺动脉及其分支形态正常,肺循环时间(pulmonary circulation time,PCT)为1.43 s(图1)。
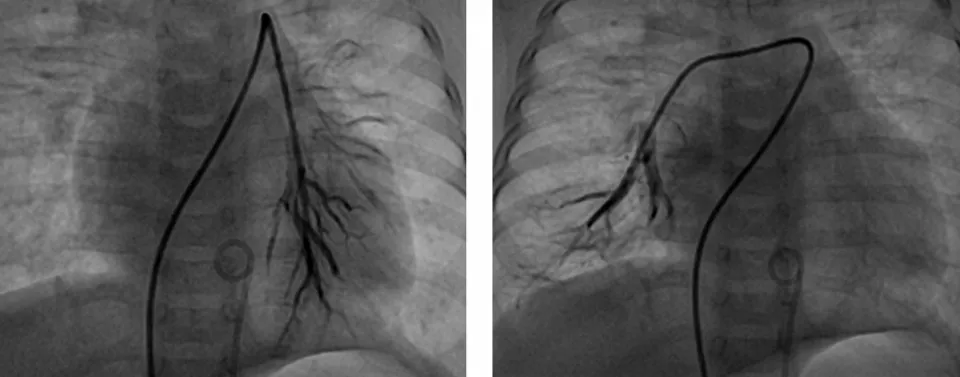
图1 室间隔缺损肺动脉高压(动力型)患儿SPA图像
患儿2的PPA:PAO=0.96,PAWP=13 mmHg,PVRI=8.0 wood·m2,AVT(-)。左、右肺动脉分支SPA 显示肺小动脉细小、轻度迂曲,呈枯树枝状,PCT 为2.85 s(图2)。

图2 室间隔缺损肺动脉高压(非动力型)患儿SPA 图像
患儿3的PPA:PAO=0.69,PAWP=15 mmHg,PVRI=4.89 Wood·m2。左、右肺动脉SPA 显示左肺动脉各分支迂曲明显,毛细血管扩张,呈丛状,PCT 延长(2.14 s);右侧肺动脉分支稍迂曲,PCT 基本正常(1.67 s)(图3)。
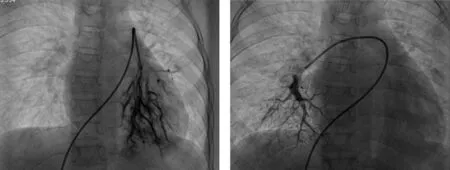
图3 IgA 肾病合并特发性肺动脉高压患儿SPA 图像
患儿4的PPA:PAO=0.85,PAWP=13 mmHg,PVRI=26.7 Wood·m2,AVT(-),左、右肺动脉SPA 显示左、右肺小动脉分支极其细小,呈毛发状,PCT 为3.56 s(图4)。
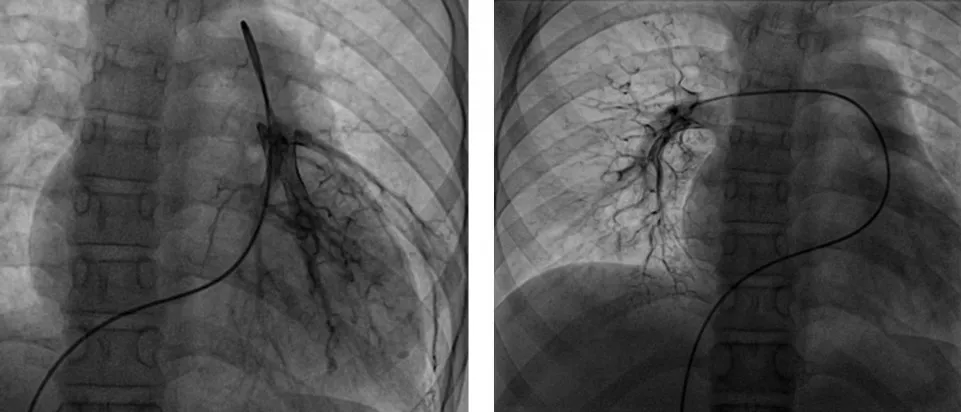
图4 BMPR2 相关性肺动脉高压SPA 图像
患儿5的PPA:PAO=0.57,PAWP=10 mmHg,PVRI=4.55 Wood·m2。吸入一氧化氮时AVT(-),吸入纯氧时AVT(+)。左、右肺动脉SPA 提示双肺动脉各分支迂曲,毛细血管扩张,呈丛状,PCT 为2.05 s(图5)。
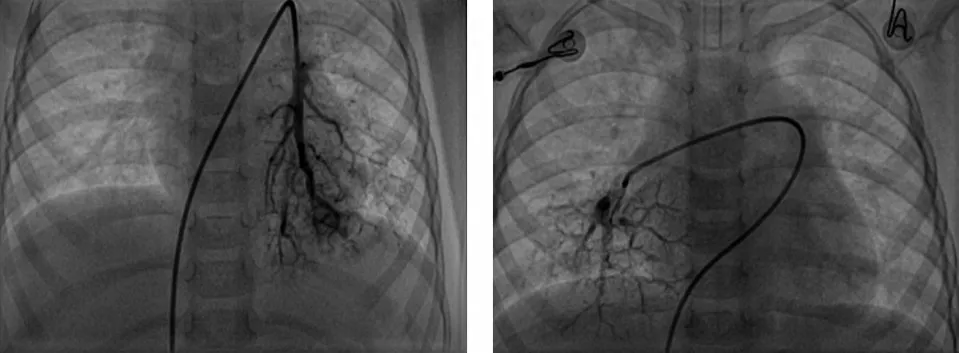
图5 ACVR1(c.789C>A,p.D263E)相关性肺动脉高压患儿SPA 图像
患儿6的PPA:PAO=0.97,PAWP=13 mmHg,PVRI=9.71 Wood·m2,吸入一氧化氮时AVT(-),吸入纯氧时AVT(+)。左肺动脉SPA 及主肺动脉造影显示双肺动脉各分支明显迂曲,毛细血管明显扩张,呈丛状,PCT 为2.56 s(图6)。

图6 ACVR1(c.1450C>T,p.R484W)相关性肺动脉高压患儿SPA 图像
2.2 患儿心导管检查血流动力学资料
6例肺动脉高压患儿心导管检查血流动力学资料总结如表1 所示。
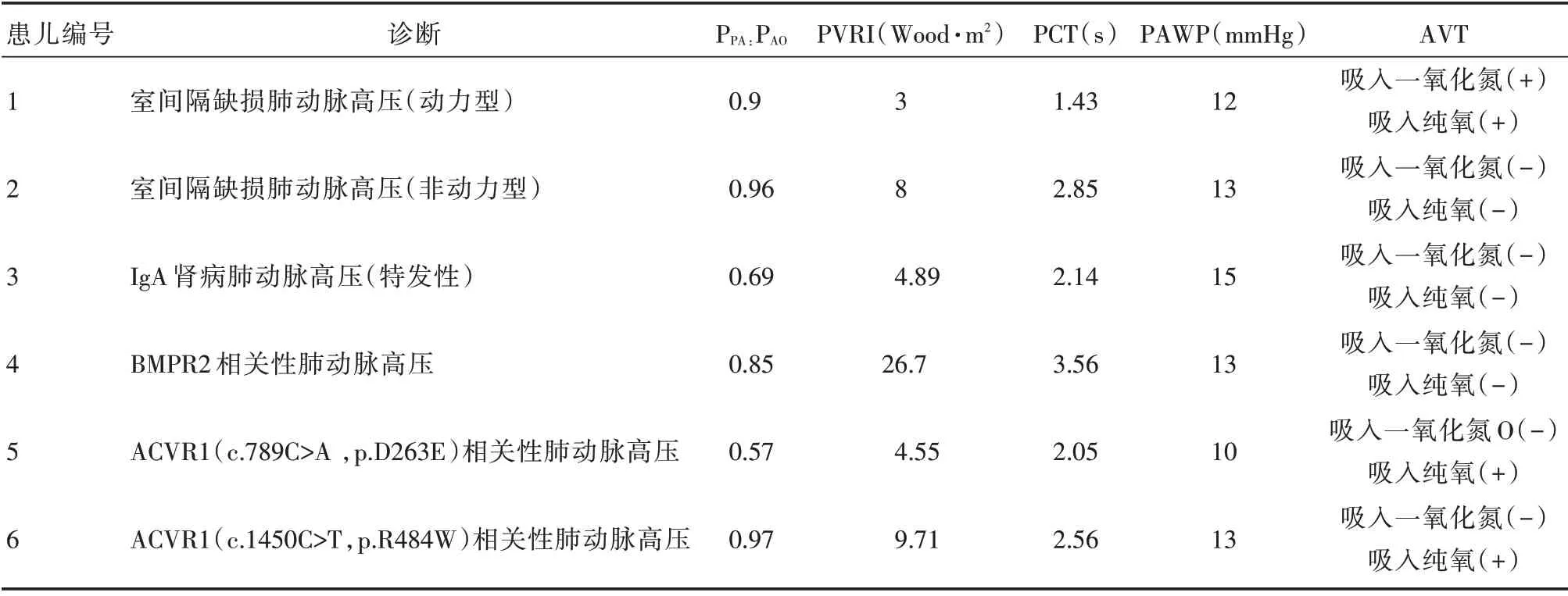
表1 不同类型患儿心导管检查血流动力学资料
3 讨论
小儿动脉型肺动脉高压按病因分为5 类,包括特发性肺动脉高压,遗传性肺动脉高压,药物及毒物所致肺动脉高压,疾病相关性肺高压如结缔组织病、人类免疫缺陷病毒感染、门脉高压、先天性心脏病、血吸虫病等,肺静脉闭塞性疾病和(或)肺毛细血管瘤样增生症,以及新生儿持续性肺动脉高压[1-3]。其中先天性心脏病导致小儿肺动脉高压为最主要的原因,而特发性肺动脉高压和遗传性肺动脉高压需要排除其他原因引起的肺高压并结合基因检查才能确诊。本文总结的6 种病例,包含先天性心脏病相关性肺高压、特发性肺高压、遗传性肺高压等,涉及不同类型肺动脉高压临床特点、心导管检查特点以及短期预后,为该类疾病的诊断和治疗提供临床经验。
先天性心脏病是导致小儿肺动脉高压最常见的原因,先天性心脏病合并肺动脉高压最主要的治疗手段是早期手术,但手术之前,必须要判断肺动脉高压性质,特别对于先天性心脏病合并重度肺动脉高压的患者。先天性心脏病相关肺动脉高压的性质主要依据患者年龄、症状以及心脏超声、胸片等来评估,但最能够反应肺动脉高压性质的仍然是心导管检查。对于左向右分流性先天性心脏病,PVRI<6 Wood·m2可以直接进行手术;PVRI>8 Wood·m2,若AVT(+),可以有条件进行手术(补片中间留孔),若AVT(-),则不能进行手术,需服用靶向药物治疗至少半年,再进行心导管检查评估,若AVT(+),则有条件手术,若仍AVT(-),则可能不能再进行先天性心脏病手术。以上两种情况在临床治疗上比较好决断,相对棘手的是6 Wood·m2<PVRI>8 Wood·m2的灰色地带,需要综合判断,除了结合AVT 结果外,部分外科医生比较看重SPA 结果,若AVT(+),肺小动脉无枯树枝状,则可进行有条件甚至根治手术,反之,若AVT(-),肺小动脉狭窄呈枯树枝状,则需进行靶向药物治疗后再评估能否进行手术[6-10]。本文患儿1 的PVRI 为3 Wood·m2,肺小动脉形态正常,PCT 正常,因此考虑动力型肺动脉高压,直接进行室间隔缺损根治术,而患儿2的PVRI为8 Wood·m2,肺小动脉狭窄、迂曲,呈枯树枝状,PCT 延长,AVT(-),考虑为非动力型肺动脉高压,无手术适应证,行口服靶向药物治疗。
患儿3 为IgA 肾病治疗期间偶然发现肺动脉高压,经多方寻找未发现肺动脉高压原因,也因此影响原发病的治疗。在进行心导管检查后,发现患儿左肺动脉分支迂曲明显,毛细血管扩张,呈丛状,PCT 延长,考虑左肺动脉血管畸形。结合患儿有习惯性鼻衄、肝血管瘤病史,高度怀疑有遗传性毛细管增生症可能。但基因检查未发现明确相关致病基因,最终考虑特发性肺高压,环磷酰胺和糖皮质激素加重肺高压病情,具体机制不详,目前国内外暂无类似病例报道。患儿4、5、6 均为基因相关性肺动脉高压,是目前报道中最常见的两种基因,即BMPR2 和ACVR1。BMPR2与肺动脉肌层及内膜层生成有关,是报道中引起遗传性相关肺动脉高压最常见的基因[11-12]。ACVR1 常见于遗传性毛细血管扩张症[13],其中有20%可以导致肺动脉高压。多篇文献报道BMPR2和ACVR1 相关肺动脉高压相对特发性肺高压预后较差,其中,BMPR2 预后最差[14-16]。本文的BMPR2相关性肺动脉高压(患儿4)患儿的肺小动脉发育明显细小,肺动脉阻力在所有患儿中最大,AVT 对一氧化氮及纯氧均无反应,临床治疗效果不明显,似乎能够解释该肺动脉高压预后较差的原因。而ACVR1 相关性肺动脉高压SPA 也具有一定的特点,即细小肺动脉扩张、迂曲,成丛状,伴有PCT延长。两例ACVR1 相关性肺炎患儿吸入纯氧时AVT(+),给予间断氧疗联合波生坦靶治疗,效果优于BMPR2 相关性肺炎,和报道一致。
综上所述,心导管检查作为肺动脉高压诊断的“金标准”,在治疗及预后上也具有一定指导价值,特别是SPA 对肺动脉高压分型具有一定诊断意义。
