川明参粗多糖初级结构解析及其体外抗氧化活性
2021-09-09唐华丽罗振宇罗黄洋
高 涛,唐华丽,罗振宇,罗黄洋,李 敏
(重庆三峡学院生物与食品工程学院 重庆 404100)
植物多糖作为一类大分子化合物,具有抗肿瘤[1]、抗病毒[2]、抗氧化[3]、降血糖[4]、增强免疫等生物活性[5]。川明参(Chuanminshen violaceum Sheh et Shan)属伞形科多年生草本植物,主要分布在四川和湖北。作为传统中药,川明参具有止咳化痰、滋阴养胃、健脾的功效[6-7]。据报道,川明参多糖对遗传物质损伤也具有一定的修复作用[8];能显著提高超氧化物歧化酶 (Cu/Zn-SOD,Mn-SOD,Fe-SOD)、过氧化氢酶(CAT)、谷胱甘肽过氧化物酶(GSH-Px)和硫氧还蛋白(Trx)的mRNA 表达水平[9];通过促进淋巴细胞增殖显著提高环磷酰胺诱导的小鼠的免疫活性,以及作为佐剂提高口蹄疫病毒疫苗免疫应答[10-11]。硫酸化川明参多糖不仅具有治疗自身免疫性疾病和免疫抑制类疾病的潜力,还具有抗鸭肠炎病毒的活性[11-12]。此外,硒化川明参多糖作为佐剂能增强乙肝疫苗的免疫应答[13]。
自由基是指含有未配对电子的基团、分子或原子,其包括超氧阴离子(O2-)、羟自由基(·OH)、脂质过氧化物(LPO)等,它是人体内正常代谢的产物。一般情况下,人体内的自由基水平处于动态平衡中,平衡一旦被打破,由于其状态极不稳定,其会攻击邻近分子(碳水化合物、蛋白质、脂质等)以夺取电子。过多的自由基会导致细胞结构破坏、功能丧失,造成细胞损伤或死亡[14-16]。
本研究通过1-苯基-3-甲基-5-吡唑啉酮(PMP)柱前衍生结合超高效液相色谱-质谱连用(UPLC-MS/MS)分析川明参粗多糖的单糖组成;采用高效凝胶渗透色谱(HPGPC)分析其分子质量分布;通过紫外-可见光光谱(UV)、傅里叶红外光谱(FT-IR)以及核磁共振光谱(NMR)鉴定川明参粗多糖结构;通过羟自由基清除率、Fe2+螯合能力以及还原力试验研究川明参粗多糖的体外抗氧化能力,为进一步开发川明参多糖提供一定依据。
1 材料与方法
1.1 材料与试剂
干制川明参购自重庆市万州区中药材批发市场,经中药粉碎机粉碎。
葡萄糖(Glc),甘露糖(Man),半乳糖(Gal),核糖(Rib),岩藻糖(Fuc),木糖(Xyl),鼠李糖(Rha),阿拉伯糖(Ara),古洛糖醛酸(Gudo),葡萄糖醛酸(GlcA),甘露糖醛酸(ManA),半乳糖醛酸(GalA),氨基半乳糖(GalN),氨基葡萄糖GlcN),美国sigma 公司;窄分布聚乙二醇,美国Polymer Standards Service 公司;牛血清蛋白、Folin 酚试剂,福州飞净生物科技有限公司;硫酸亚铁、过氧化氢、铁氰化钾、菲洛嗪,国药集团化学试剂有限责任公司;苯酚、浓硫酸、1-苯基-3-甲基-5-吡唑啉酮(PMP)等均为国产分析纯级,西陇科学股份有限公司。
1.2 仪器与设备
KQ-300B 超声清洗仪,昆山市超声仪器有限公司;UV-2450 紫外-可见光分光光度计,日本岛津公司;FTIR-650 傅里叶变换红外光谱仪,天津港东科技股份有限公司;ALPH1-2/LD-Plus 冷冻干燥机,德国CHRIST 公司;液相色谱-质谱联用仪 (I-class UPLC,Xevo TQ-S micro 三重四级杆质谱)、1515 系列高效液相色谱,美国Waters 公司;Bruker AVance III 400MHz 核磁共振波谱仪,德国Bruker 公司。
1.3 方法
1.3.1 粗多糖提取与纯化 称取一定量川明参粉置于2 000 mL 锥形瓶中,按料液比1∶20 加入蒸馏水溶解。在300 Hz、60 ℃条件下,将溶解液放入超声清洗仪中超声提取90 min。超声完成后用4层纱布过滤,然后将滤液浓缩至原体积的1/4,向浓缩液中加入无水乙醇,直至乙醇的体积分数为80%,静置沉淀,过夜后离心收集沉淀,经真空冷冻干燥得到粗多糖粉末。称取一定量粗多糖粉末配制成10 mg/mL 溶液,采用Sevag 法除去游离蛋白。按体积比3∶1 的比例向川明参粗多糖溶液加入氯仿-甲醇(体积比4∶1)溶液,剧烈震荡30 min后离心,收集上清液后用分液漏斗收集上层液体。重复6 次后按上述步骤加入无水乙醇提取川明参粗多糖并真空干燥得到川明参粗多糖。
1.3.2 川明参粗多糖结构鉴定
1.3.2.1 单糖组成测定 采用PMP 柱前衍生UPLC-MS/MS 法测定单糖组成[17-19]。称取10 mg 多糖样品置于20 mL 安瓿瓶中,加入5 mL 三氟乙酸(2 mol/L)溶液,N2封管,100 ℃条件下水解2 h。水解完成后取1 mL 水解液加入1 mL 甲醇于70 ℃条件下用N2吹干,重复2 次以去除三氟乙酸。加入1 mL NaOH(0.3 mol/L)溶解残渣。分别取400 μL 的样品水解液与单糖标准溶液于5 mL 具塞试管中,加入400 μL 的PMP 甲醇溶液,漩涡混匀,于70 ℃下反应2 h,反应完成后去除放置至室温,再加入400 μL HCl(0.3 mol/L)调节pH 值至6~7,加水1 200 μL,再加入等体积的氯仿萃取,涡旋混匀,静置后收集水相,重复2 次。将水相用0.45 μm 水系滤膜过滤后供UPLC-MS/MS 分析。
HPLC 条 件 为Agilent EC-C18 色谱柱(2.1 mm×50 mm,2.7 μm);流量0.4 mL/min;流动相A:20 mmol/L 乙酸铵缓冲液 (pH=7);流动相B:乙腈;梯度洗脱模式:时间梯度为0 min-1 min-7 min-11 min-13 min-14.5 min-14.6 min-17 min,流动相B 的体积分数梯度为14%-14%-18.5%-20%-60%-60%-14%-14%。
质谱条件为ESI+模式;喷雾电压2.0 kV;锥孔电压30 V;离子源温度150 ℃;脱溶剂温度500℃;脱溶剂气体N2,流速为1 000 mL/h;质谱扫描范围:170~800 m/z。
1.3.2.2 分子质量测定 分子质量测定采用HPGPC 法。取样品5 mg 加入5 mL 试管中,加入超纯水配置的0.1 mol/L 硝酸钠溶液,超声溶解后过0.22 μm 水系滤膜上HPLC 分析。以分子质量分别为330 000,176 000,82 500,44 000,25 300,20 600,12 600,7 130,4 290,1 400,633,430 的窄分布聚乙二醇为对照。
色谱条件为ULTRAHYDROGEL 120 PKGD,ULTRAHYDROGEL 250 PKGD,ULTRAHYDROGEL 500 PKGD 色谱柱;Waters 2414 RID 检 测器,流动相:0.1 mol/L 硝酸钠;流量1 mL/min。
1.3.2.3 β 消去反应 称取0.1 g 干燥的川明参粗多糖,溶于100 mL NaOH 溶液(0.2 mol/L)中,于45 ℃水浴3 h,在波长200~500 nm 范围内进行紫外光谱扫描。以蒸馏水代替NaOH 溶液作为空白对照。
1.3.2.4 傅里叶红外光谱(FT-IR) 称取1~2 mg干燥后的川明参粗多糖粉末,置于玛瑙研钵中,加入150~200 mg 干燥后的KBr 晶体,研磨均匀后压片,在4 000~400 cm-1范围测定其红外光谱。
1.3.2.5 核磁共振光谱(NMR) 称取适量川明参粗多糖样品溶解于99.9% D2O 中,并使用Bruker 400 MHz 核磁共振波谱仪测定样品的1H-NMR 和13C-NMR。
1.3.3 体外抗氧化试验
1.3.3.1 总还原力测定 取1.3 mL 不同质量浓度的样品溶液,分别加入pH=6.6 的磷酸盐缓冲液1.3 mL,1%铁氰化钾溶液0.5 mL,混合均匀后于50 ℃条件下水浴25 min,快速冷却后加入10%的三氯乙酸溶液1.3 mL,充分混合后于6 000 r/min条件下离心10 min。取上清液1.3 mL,加入1.3 mL蒸馏水和0.1%的三氯化铁溶液0.3 mL,摇匀后反应10 min,于波长700 nm 处测定吸光度。每组平行测定3 次,取平均值。
1.3.3.2 羟自由基清除率的测定 取2.2 mL 不同质量浓度的样品溶液,加入9 mmol/L 硫酸亚铁溶液2.2 mL,9 mmol/L 水杨酸2.2 mL 和8.8 mmol/L过氧化氢溶液2.2 mL,充分摇匀后置于37 ℃条件下水浴25 min,冷却后于波长510 nm 处测定其吸光度。每组平行测定3 次,取平均值。羟自由基清除率计算公式如下:
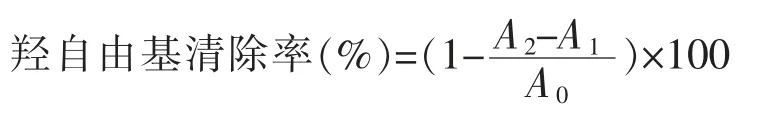
式中,A0——去离子水代替样品溶液反应后的吸光值;A1——去离子水代替过氧化氢溶液反应后的吸光值;A2——样品溶液反应后的吸光值。
1.3.3.3 Fe2+螯合能力测定 取1.0 mL 不同质量浓度的样品溶液溶液,加入蒸馏水3.0 mL 和2.0 mmol/L 氯化亚铁溶液0.1 mL,充分混合后加入5.0 mmol/L,菲洛嗪溶液0.2 mL,充分反应10 min后于波长562 nm 处测定溶液吸光度值。每组平行测定3 次,取平均值。Fe2+螯合能力计算公式如下:
式中,A0——去离子水代替样品溶液反应后的吸光值;A1——去离子水代替氯化亚铁溶液反应后的吸光值;A2——样品溶液反应后的吸光值。

2 结果与分析
2.1 单糖组成及分子质量分析
利用PMP 柱前衍生UPLC-MS/MS 法测定川明参多糖的单糖组成。标准单糖与样品的色谱图如图1所示,在m/z=525 条件下定量测定古洛糖醛酸(GulA)、葡萄糖醛酸(GlcA)、甘露糖醛酸(ManA)和半乳糖醛酸(GalA);在m/z=511 条件下定量测定葡萄糖(Glc)、甘露糖(Man)和半乳糖(Gal);在m/z=510 条件下定量测定氨基半乳糖(GalN)和氨基葡萄糖(GlcN);在m/z=495 条件下定量测定鼠李糖(Rha)和岩藻糖(Fuc);在m/z=481 条件下定量测定核糖(Rib)、木糖(Xyl)和阿拉伯糖(Ara)。采用峰面积归一法计算川明参粗多糖的单糖组成,结果表明,川明参粗多糖主要由鼠李糖(1.24%)、半乳糖醛酸(1.81%)、核糖(3.47%)、半乳糖(8.42%)、甘露糖(17.67%)、阿拉伯糖/木糖(27.83%)和葡萄糖(39.55%)组成。此外,川明参粗多糖的HPGPC 色谱图如图2所示,其结果表明,川明参粗多糖是一种分子质量11.7 ku 的多分枝状多糖。
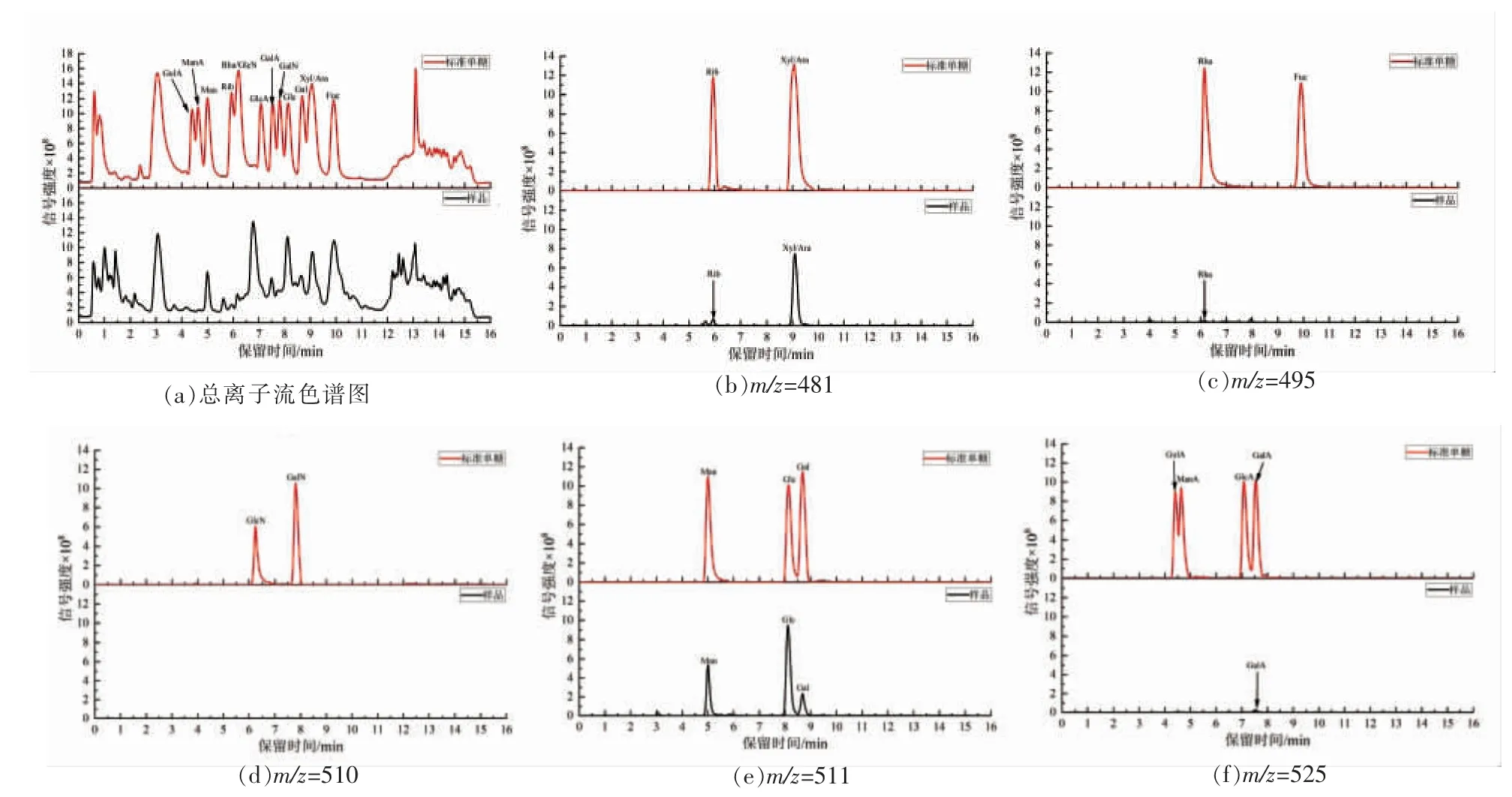
图1 PMP 柱前衍生UPLC-MS/MS 色谱图Fig.1 UPLC-MS /MS chromatogram derived before PMP column

图2 川明参粗多糖的HPGPC 色谱图Fig.2 HPGPC chromatogram of crude polysaccharides from C.violaceum
2.2 川明参粗多糖中糖肽键的连接方式
β 消去反应是测定糖蛋白中糖肽键连接方式的传统方法。-O-型糖苷键与糖链连接的苏氨酸与丝氨酸在低碱条件下会水解生成α-氨基丙烯酸与α-氨基丁烯酸,可引起波长240 nm 处的吸光度值升高[20-22]。如图3所示,在NaOH 处理后,川明参粗多糖的紫外光谱在波长240 nm 处有明显上升,表明川明参多糖为-O-型糖肽键连接的蛋白聚糖。

图3 样品的全扫描紫外光谱图Fig.3 Full scan ultraviolet spectrum of sample
2.3 官能团分析
川明参粗多糖红外光谱如图4所示,3 410 cm-1处较宽的吸收峰由分子间和分子内O-H 和N-H 的伸缩振动引起的;2 930 cm-1处的吸收峰为多糖的C-H 伸缩振动峰,此为多糖的特征吸收峰;1 640 cm-1处的吸收峰为一级氨基和二级氨基的N-H 的边角振动或羰基-C=O 的非对称伸缩振动;1 415 cm-1处的吸收峰为多糖的C-O 和蛋白质的C-N 伸缩振动;1 000~1 200 cm-1范围内的3个吸收峰表明川明参粗多糖主要由吡喃糖构成;920 cm-1和850 cm-1处的吸收峰表明川明参粗多糖既含有α-吡喃糖苷键,又有β-吡喃糖苷键。
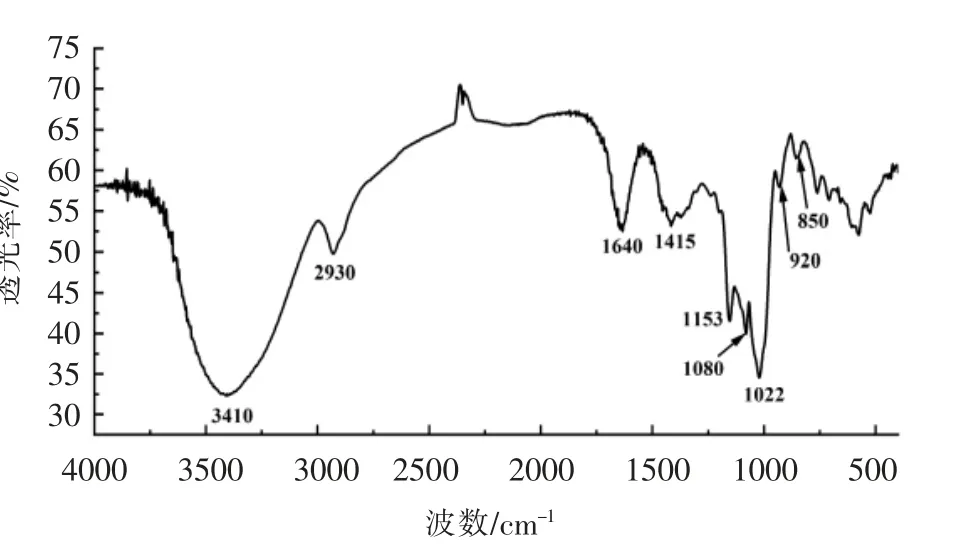
图4 样品的傅里叶红外光谱图Fig.4 FT-IR spectrum of sample
2.4 糖残基及连接情况分析
一般而言,在异头氢质子区(δ=4.3~5.9)范围内有几个信号,表明有几种糖残基。此外,α 构型的糖残基的异头氢质子信号大于δ=5.0,β 构型的糖残基的氢质子信号小于δ=5.0[23]。川明参粗多糖的核磁共振氢谱如图5a所示,在异头质子区共出现8 个信号,分别为δ=5.32,5.31,5.26,5.13,5.06,5.00,4.88,4.56,此结果表明川明参粗多糖中含有8 种糖残基,并同时存在α 和β 构型。糖残基的H2~H6位信号出现于δ=3.0~4.2 范围内,其重合严重,难以归属。此外,在δ=3.2~3.5 范围内存在较弱的信号,表明存在较少的-OCH3;此外,在δ=1.0~1.3 范围内存在较明显的-CH3信号,根据川明参粗多糖的单糖组成结果,将其归属为甲基戊糖中的鼠李糖信号[24]。
通常而言,异头碳质子信号位于δ=90~112之间,而大多数α 构型糖残基的异头碳质子信号处于δ=90~102,β 构型糖残基的异头碳质子信号位于δ=102~112 之间[24]。川明参粗多糖的核磁共振碳谱如图5b所示,在δ=90~112 之间存在δ=107.27,104.32,103.61,99.63,99.59,98.55,95.72,92.0 8 种异头碳信号,表明川明参粗多糖中存在8 种单糖残基。此外,由于呋喃糖的C3或C5的质子信号大于δ=80,结合单糖组成与端基碳质子信号δ=107.27,图5b 中δ=81.36 可归属为α-Araf信号。此外,δ=15~20 范围内的信号也表明了鼠李糖的存在;δ=75~77 范围内的3 个信号为六碳糖的C2~C5或五碳糖的C2~C4被取代后的信号;δ=70~75 范围内的信号为C2~C5范围内未被取代的信号;δ=68~70 范围内的信号为六碳糖的C6或五碳糖的C5被取代信号;而δ=55-65 范围内的信号为未被取代的六碳糖的C6或五碳糖的C5信号。根据文献与单糖组成,可将异头质子信号δ=4.56/95.72 和δ=5.13/92.09 依次归属为→4-β-Xylp和→4-α-Xylp[25-26];δ=5.00/107.27 归属为α-1,5-Araf[27];δ=5.06/104.32 归属为α-1,2-Rhap或α-1,2,4-Rhap[28];δ=4.88/103.61 归属为β-1,4,6-Galp[29]。根据参考文献、单糖组成以及C 谱中较明显信号,将δ=5.32/99.636 和δ=5.31/99.59 归属于α-1,4-Glc,α-1,6-Glc 或α-1,4,6-Glc[29-30];δ=5.26/98.55 应归属为α-1,2-Man[31-32]。综上所述,川明参粗多糖中主要为α-1,4-糖苷键。此外,还含有一定量的α-1,2-糖苷键、α-1,6-糖苷键和α-1,5-糖苷键以及极少量的β-1,4-糖苷键和β-1,6-糖苷键。
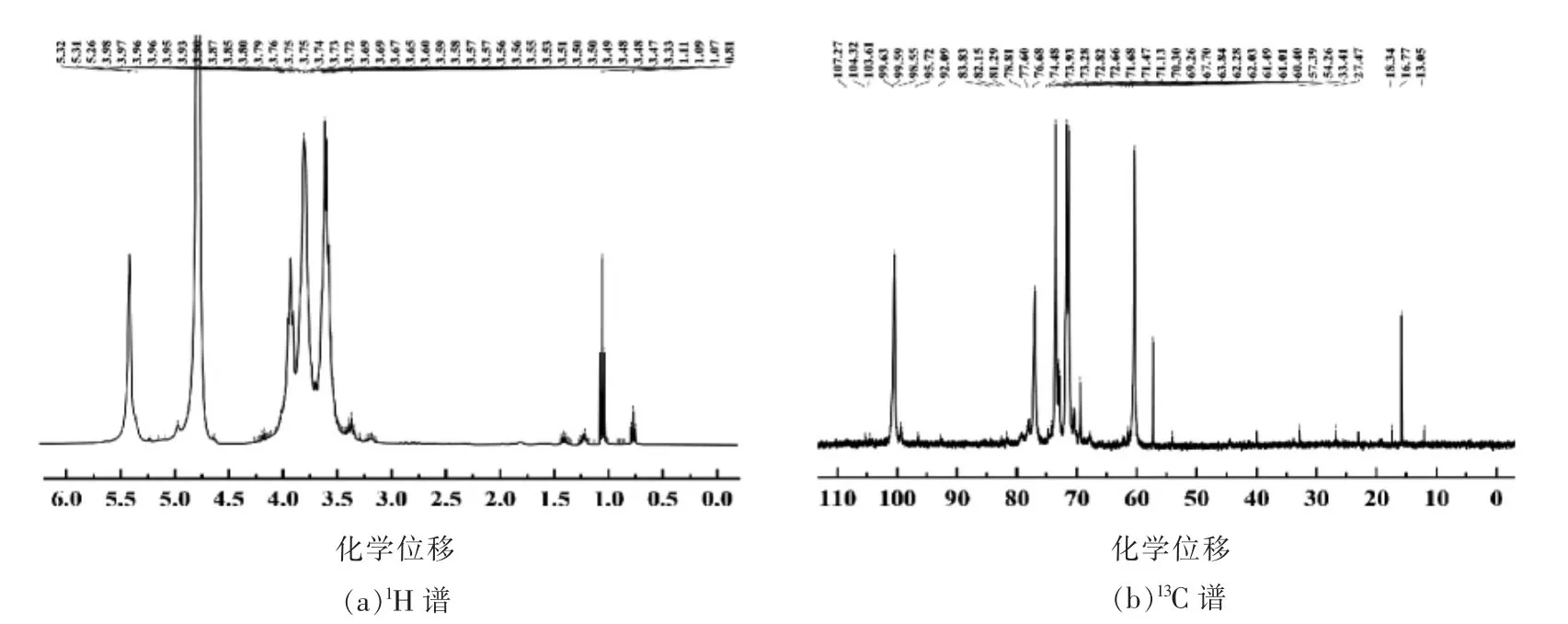
图5 样品的核磁共振光谱Fig.5 NMR spectra of samples
2.5 川明参粗多糖的体外抗氧化活性
羟自由基是一类化学性质非常活跃的自由基,易与碳水化合物、蛋白质、脂质和DNA 发生反应,导致细胞损伤或死亡[33]。川明参粗多糖羟自由基清除率如图6a所示,在低质量浓度(0~1 g/mL)范围,川明参粗多糖的羟自由基清除率与质量浓度存在明显的剂量效应关系;当质量浓度大于1 g/mL 后,羟自由基清除率无明显增加。虽然川明参粗多糖的羟自由基清除率显著低于VC,但质量浓度在1 g/mL 时,其羟自由基清除率为(60.17±1.96)%,表明其具有较好的羟自由基清除能力。
金属离子可通过向化合物转移单个电子从而引发自由基的形成,所以,Fe2+螯合能力与抗氧化活性密切相关[34]。如图6b所示,川明参粗多糖的Fe2+螯合能力在低质量浓度(0~1 g/mL)范围具有较好的剂量效应关系,当质量浓度大于1 g/mL时,其Fe2+螯合能力无明显增加,此时的Fe2+螯合能力为(63.2±1.39)%,表明其具有较好的抑制自由基形成的能力。
川明参粗多糖的总还原力如图6c所示,其在低质量浓度(0~0.6 g/mL)范围内有明显的剂量效应关系;当质量浓度大于0.6 g/mL 时,其吸光度值无明显增加。结果表明,川明参粗多糖具有一定的还原力,但其还原力低于VC。
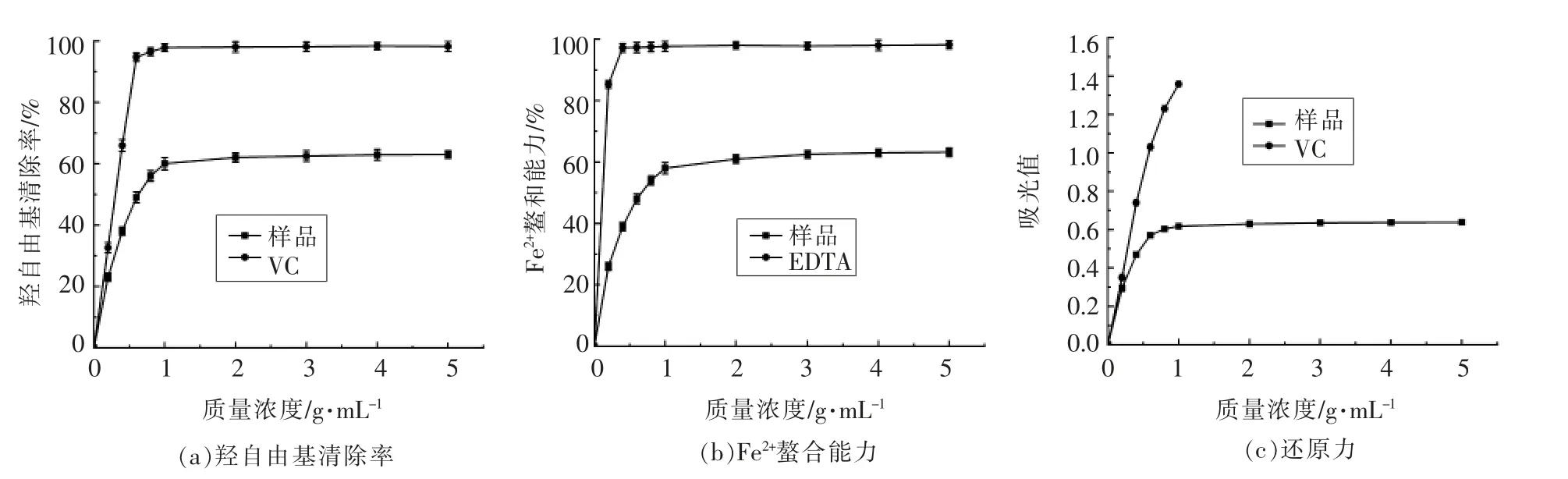
图6 样品的抗氧化试验结果Fig.6 Results of antioxidant test of the samples
3 结论
通过PMP 柱前衍生结合UPLC-MS/MS 分析得到,川明参粗多糖主要由由鼠李糖(1.24%)、半乳糖醛酸(1.81%)、核糖(3.47%)、半乳糖(8.42%)、甘露糖(17.67%)、阿拉伯糖/木糖(27.83%)和葡萄糖(39.55%)组成;通过HPGPC 分析得到,川明参粗多糖为分子质量为11.7 ku 的多分枝状多糖;通过β 消去反应得到,川明参粗多糖中具有-O-连接的蛋白聚糖;红外光谱分析表明,川明参粗多糖中α-吡喃糖苷键与β-吡喃糖苷键同时存在;最后通过NMR 分析得到,川明参粗多糖中主要为α-1,4-糖苷键;此外,还含有一定量的α-1,2-糖苷键、α-1,6-糖苷键和α-1,5-糖苷键以及极少量的β-1,4-糖苷键和β-1,6-糖苷键。抗氧化试验结果表明,虽然川明参粗多糖的羟自由基清除率、亚铁离子熬和能力和总还原力与对照品还存在一定差距,但在低质量浓度范围内,其羟自由基清除率,亚铁离子熬和能力和总还原力都存在明显的剂量效应关系,当川明参粗多糖质量浓度较高时,其表现出明显的抗氧化活性。本研究为进一步开发川明参多糖提供一定的依据。
