发芽糙米多糖的结构解析及降糖活性分析
2021-09-09刘晓飞马京求林枞雨卢淑雯
刘晓飞,马京求,侯 艳,张 宇,林枞雨,张 娜*,卢淑雯
(1 哈尔滨商业大学食品工程学院 哈尔滨 150076 2 哈尔滨工业大学生命科学与技术学院 哈尔滨 150080 3 黑龙江省农业科学院博士后科研工作站 哈尔滨 150086)
发芽糙米 (Germinated brown rice,GBR)是由稻谷脱壳后不加工或较少加工的全谷粒米在适宜的水分、温度等外界条件下发芽所得,发芽后的糙米细胞组织容易软化,便于煮熟[1]。在加工过程中GBR 中原有的蛋白质和淀粉等物质易被降解,这使其口感和风味都得到提高,而且GBR 中保留了原有的丰富维生素、矿物质(Zn、Mg、K、Fe)等营养物质,还生成了多种像γ-氨基丁酸 (γ-Aminobutyric acid,GABA)等有利于人体健康的物质[2-4]。这些成分使GBR 不仅具有较高的营养价值,还具有各种功能活性[5],是一种新兴的“医食同源”产品[6]。
多糖是GBR 中的主要成分之一[7]。随着时代的发展,对于活性多糖的研究近年来越来越多[8]。然而,目前对GBR 的研究主要集中在发芽的工艺研究[9]、γ-氨基丁酸富集[10]、淀粉的改性[11]以及以GBR 为原料的各种食品制作上[12-13]。有关发芽糙米多糖(Germinated brown rice polysaccharides,GBRPs)的结构分析以及功能活性还未见报道。
本研究采用水提醇沉法、酶-Sevage 法及大孔树脂吸附法提取、纯化GBRPs,对脱蛋白、脱色处理后的GBRPs 进行柱层析分离,分离出不同组分的GBRPs,再通过紫外-可见光光谱法、凝胶色谱法、高效液相色谱法、傅里叶红外光谱、核磁共振氢谱法分别测定各组分GBRPs 的分子质量、单糖组成、化学键组成、糖链结构,通过细胞试验证实其多糖组分具有降糖的活性作用,为进一步研究发芽糙米多糖更深层次的作用机制、精细结构,开发有利于人体健康的功能性食品提供理论依据。
1 材料与方法
1.1 材料与试剂
1.1.1 发芽糙米多糖样品制备 供试粳稻为 “龙粳”,用龚谷机去除稻子外壳得到糙米,在4 ℃避光的条件下,将糙米浸泡在无菌水中12 h。取出糙米后放置在潮湿的环境中,在20 ℃条件下培养过夜。去除掉没有发芽的糙米后放置在50 ℃恒温干燥箱中干燥24 h,粉碎过筛。按料液比1∶30 加入蒸馏水,搅拌过夜,取上清液并浓缩,按体积比1∶3 加入85%乙醇,静置24 h,去除掉上清液,将沉淀冷冻干燥后得发芽糙米粗多糖[14-15]。
1.1.2 试剂 DEAE Sepharose CL-6B、Sepharose CL-6B、Sephadex G50,Pharmacia 公司;无水乙醇、氯仿、浓硫酸(均为分析纯级),天津博迪化工股份有限公司;鼠李糖(Rha)、半乳糖醛酸(GalA)、葡萄糖(Glc)、甘露糖(Man)、木糖(Xyl)、半乳糖(Gal)、阿拉伯糖(Ara),Sigma-Aldrich(中国上海)公司;吡啶、盐酸羟胺、硼氢化钠、乙酸、甲醇等试剂均为分析纯级,购自天津基准化学试剂有限公司。
1.2 仪器与设备
ZLG-10 真空冷冻干燥机,北京博医康试验仪器有限公司;HHS-ZK4 恒温水浴锅,天津市泰斯特仪器仪表有限公司;Bruker400 核磁共振仪,瑞士Bruker 公司;2XZ (S)-2 傅里叶变换红外光谱仪,上海德英照明有限公司;PROSTAR21 高效液相色谱仪,美国瓦里安科技有限公司。
1.3 方法
1.3.1 GBRPs 的纯化与分离
1.3.1.1 酶-Sevage 法脱蛋白 在37 ℃条件下,向10 mg/mL 多糖溶液中加入2%的木瓜蛋白酶,酶解24 h。然后升温至80 ℃使酶失活,离心,上清液经Sevage 法脱蛋白,反复多次,直到无明显的蛋白产生为止[16],根据1.3.2 节计算脱蛋白率及多糖损失率。
1.3.1.2 大孔树脂吸附法脱色 由于发芽糙米多糖为弱极性,所以本研究选用AB-8 大孔树脂为填料,湿法装柱,柱下端塞入脱脂棉并压平,待柱内树脂稳定后,将4 BV 的2 mg/mL 的粗多糖溶液以2 BV 的速度进行脱色,待样品液完全进入柱内后,用2 倍量蒸馏水进行冲洗,收集脱色后液浓缩至上样体积[16],根据1.3.2 节计算脱色率及多糖损失率。
1.3.1.3 GBRPs 的柱层析分离 采用DEAE Sepharose CL-6B 柱层析对脱蛋白和脱色后的发芽糙米多糖进行柱层析分离[16],用苯酚-硫酸跟踪检测含糖管A490nm,绘制梯度洗脱曲线,按照洗脱曲线收集各个主峰,Sephadax G50 脱盐柱进行脱盐,浓缩后真空冷冻、干燥备用。
为提高多糖的均一性,继续用凝胶柱对经过柱层析分级分离的多糖进行再次纯化。采用湿法装柱(Φ 26 mm×600 mm)的方法,将分离的各个组分多糖溶于蒸馏水中,将2 mL 上清液上柱,用0.1 mol/L NaCl 以1 mL/min 的流速洗脱,每管收集2 mL 洗脱液,采用苯酚-硫酸法跟踪检测含糖管,绘制洗脱曲线,收集各个主峰,Sephadax G50 脱盐柱进行脱盐,浓缩后真空冷冻干燥备用。
1.3.2 计算公式

1.3.3 GBRPs 不同组分的结构分析
1.3.3.1 纯度及分子质量的测定 采用紫外-可见光谱分析(UV-Vis)方法[17],用蒸馏水充分溶解2 mg 发芽糙米多糖组分配制成多糖溶液,在紫外-可见光波长范围内进行检测。采用凝胶渗透色谱分析(GPC)方法[18]检测GBRPs 不同组分的分子质量,准确称量各组分GBRPs 1 mg,溶于1 mL 去离子水中,采用凝胶渗透色谱检测分子质量。GPC 色谱仪型号:LC-10A型HPLC,检测器:示差检测器RID-10A,进样器:自动进样器SIL-10AD,系统软件:SHIMADZU CLASS-VP,色谱柱:UltrahydrogelTMLinear 7.8 mm×300 mm。试验条件:流动相为超纯水,流速0.5 mL/min,柱温40 ℃,柱操作压力1.6 MPa,样品质量浓度1 mg/mL,进样量20 μL。
1.3.3.2 单糖组成分析 称取2 mg 样品于三角瓶中,加入2 mL 三氟乙酸,封管,置于110 ℃烘箱中酸解7 h,冷却至室温,得到水解产物。加入0.3 mL蒸馏水溶解,调节pH 值至中性,用水定容至1 mL 备用。单糖对照品为鼠李糖(Rha)、木糖(Xyl)、阿拉伯糖(Ara)、甘露糖(Man)、葡萄糖(Glc)、半乳糖(Gal)。将各标准单糖样品等摩尔混合成2 mmol/L 的溶液。经纯化的GBRPs 用2 mo1/L H2SO4在100 ℃封管水解8 h,水解产物经BaCO3中和,得到的单糖样品用于后续测定[19]。流动相为V乙腈∶V水=8∶2,流速为1 mL/min,柱温为室温,进样量20 mL。
1.3.3.3 红外光谱分析 分别将1.00 mg 干燥发芽糙米多糖粉末与溴化钾(KBr)粉末混合,于玛瑙研钵上研磨均匀,压片5 min 后,将样品在波长4 000~400 cm-1范围进行光谱扫描[20]。
1.3.3.4 一维核磁共振氢谱检测 分别取GBRPs各个组分样品20 mg 经D2O 交换3 次后,溶于0.5 mL D2O 中,用BRUKER 400 核磁共振仪,测定1H NMR (频率400 MHz,温度296.9 K)[21]。
1.3.4 GBRPs 的体外降血糖活性
1.3.4.1 GBRPs 不同组分的α-葡萄糖苷酶抑制活性 不同GBRPs 组分样品配置成质量浓度为0.5 mg/mL 的溶液,以阿卡波糖为对照组,配置浓度为0.05 mol/L 的磷酸盐缓冲液,调节缓冲液pH值稳定在6.8,分别取出0.60 mL 磷酸缓冲液和0.20 mL 的α-葡萄糖苷酶溶液(0.2 U/mL)与0.10 mL GBRPs 的不同组分溶液混合作为待测样品;将混合溶液在37 ℃水浴10 min 后加入浓度为20 mmol/L 的对硝基苯基-β-D-吡喃葡萄糖苷溶液0.20 mL,5 min 后加入1 mL 1 mol/L 的Na2CO3溶液终止该反应[22]。于波长405 nm 处测其吸光值,计算不同组分GBRPs 对α-葡萄糖苷酶的抑制率,筛选出对α-葡萄糖苷酶抑制作用最明显的多糖组分进行后续研究,抑制率计算公式如下所示:
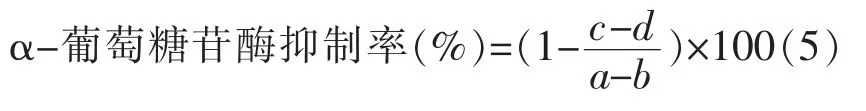
其中:a——不含样品的α-葡萄糖苷酶溶液吸光光度值;b——既不含有样品也不含α-葡萄糖苷酶溶液的吸光光度值;c——含有样品的α-葡萄糖苷酶混合溶液的吸光光度值;d——不含α-葡萄糖苷酶的样品吸光光度值。
1.3.4.2 GBRPs 不同组分对胰岛素抵抗HepG2 细胞糖代谢的影响 配制浓度为0.05 mol/L 的Tris-HCl 溶液和磷酸盐缓冲液(PBS),调节PBS 的pH值到7。将细胞取出加入离心管中,1 000 r/min 离心10 min,收集沉淀。加入0.7 mL 的PBS,轻轻将其混匀,1 000 r/min 离心10 min,收集沉淀[23]。采用超声细胞破碎仪对细胞进行破碎,超声功率为300 W,超声时间为4 s,反复对细胞进行超声破碎操作4 次,每次间隔30 s,超声过程在冰水浴中进行。分别按照丙酮酸激酶(Pyruvate kinase,PK)试剂盒和己糖激酶(Hexokinase,HK)试剂盒的说明书进行检测[24]。重复上述试验操作步骤,将细胞裂解液对细胞进行裂解,每管中加入NaOH 溶液0.25 mL 并沸水浴20 min,冷却到室温,加入双蒸水0.25 mL,按照糖原试剂盒说明书对HepG2 细胞的糖原含量进行测试,每个样品做3 个平行试验。
2 结果与分析
2.1 GBRPs 的纯化分离结果
2.1.1 GBRPs 脱蛋白、脱色的结果 酶-Sevage 法去除蛋白质是通过有机溶剂将蛋白变性为沉淀,需要多次有机试剂处理才能较好的去除掉蛋白[9]。由图1可知,对GBRPs 的脱蛋白处理次数越多,多糖的损失率越大。酶-Sevage 法经过2 次处理后,脱蛋白率较大,多糖损失率较少。AB-8 大孔树脂对弱极性的有机化合物具有较强的吸附能力,对极性大的多糖类物质吸附力弱[15],由图2可知,GBRPs 经AB-8 大孔树脂吸附色素后,脱色率为86.57%,多糖损失率为28.96%。GBRPs 经两次酶-Sevage 法去除蛋白后,脱蛋白率为74.36%,多糖损失率为14.09%。
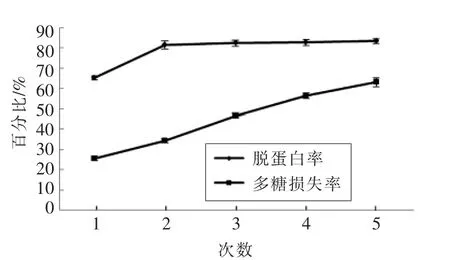
图1 酶-Sevage 法脱蛋白Fig.1 Deproteinization with the enzyme-Sevage method
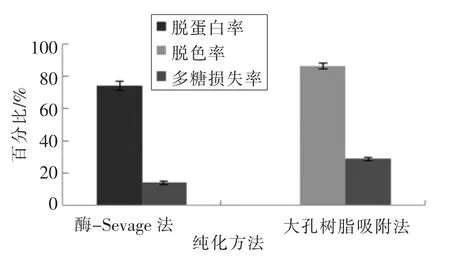
图2 脱蛋白、脱色结果Fig.2 The result of deproteinization and decolorization
2.1.2 GBRPs 柱层析分离的结果 GBRPs 层析结果如图3和图4所示。
通过图3洗脱曲线可知,GBRPs 经过阴离子层析柱分离得到4 个带电荷性质不同的洗脱峰,峰型狭窄对称,第1 个水洗多糖组分由蒸馏水洗脱得到,其含量为32.82%,经不同浓度的NaCl 梯度洗脱,得到4 个对称的明显的峰,含量分别9.91%,8.15%,7.42%和1.92%,由于0.4 mol/L 的NaCl 洗脱下来的多糖含量较少,因此只收集GBRPs 前3 个组分的洗脱液。由图4可知,水洗多糖经凝胶柱后分离得到1 个主峰,命名为WPS;含量为24.16%。盐洗多糖经凝胶柱后,收集主峰部分,得到3 个组分,分别命名为SPS-1、SPS-2、和SPS-3;含量分别为8.28%,7.09%、5.94%。
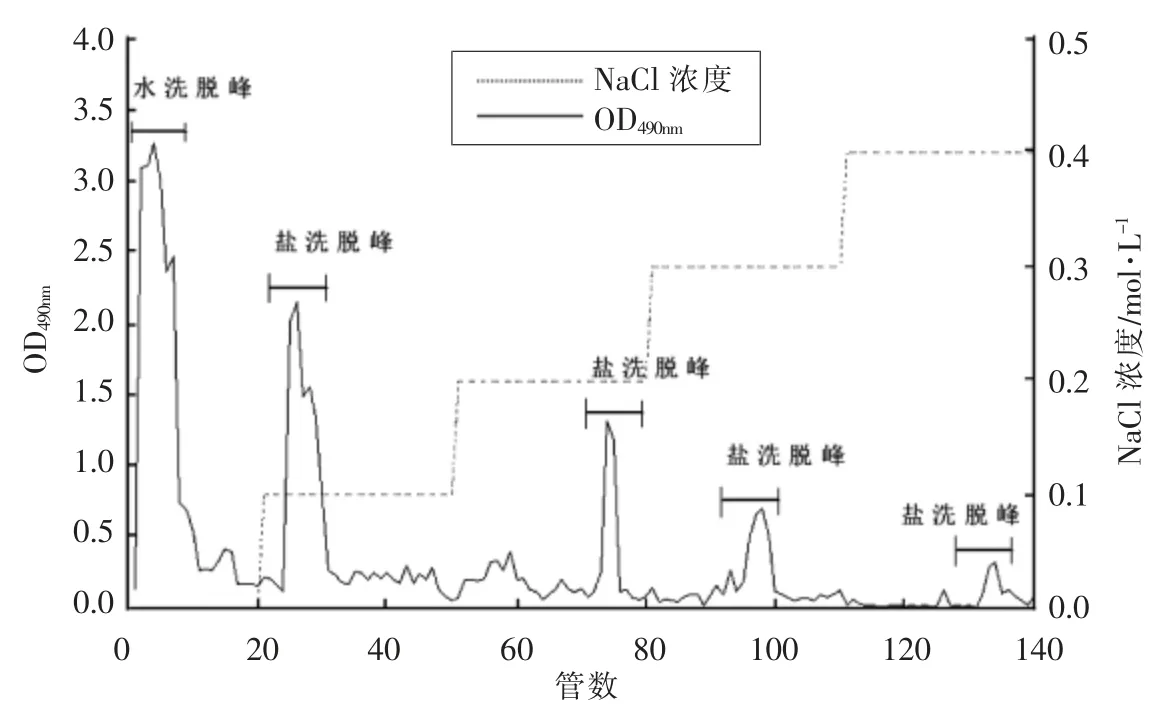
图3 DEAE Sepharose CL-6B 柱层析洗脱曲线Fig.3 DEAE Sepharose CL-6B column chromatography elution curve
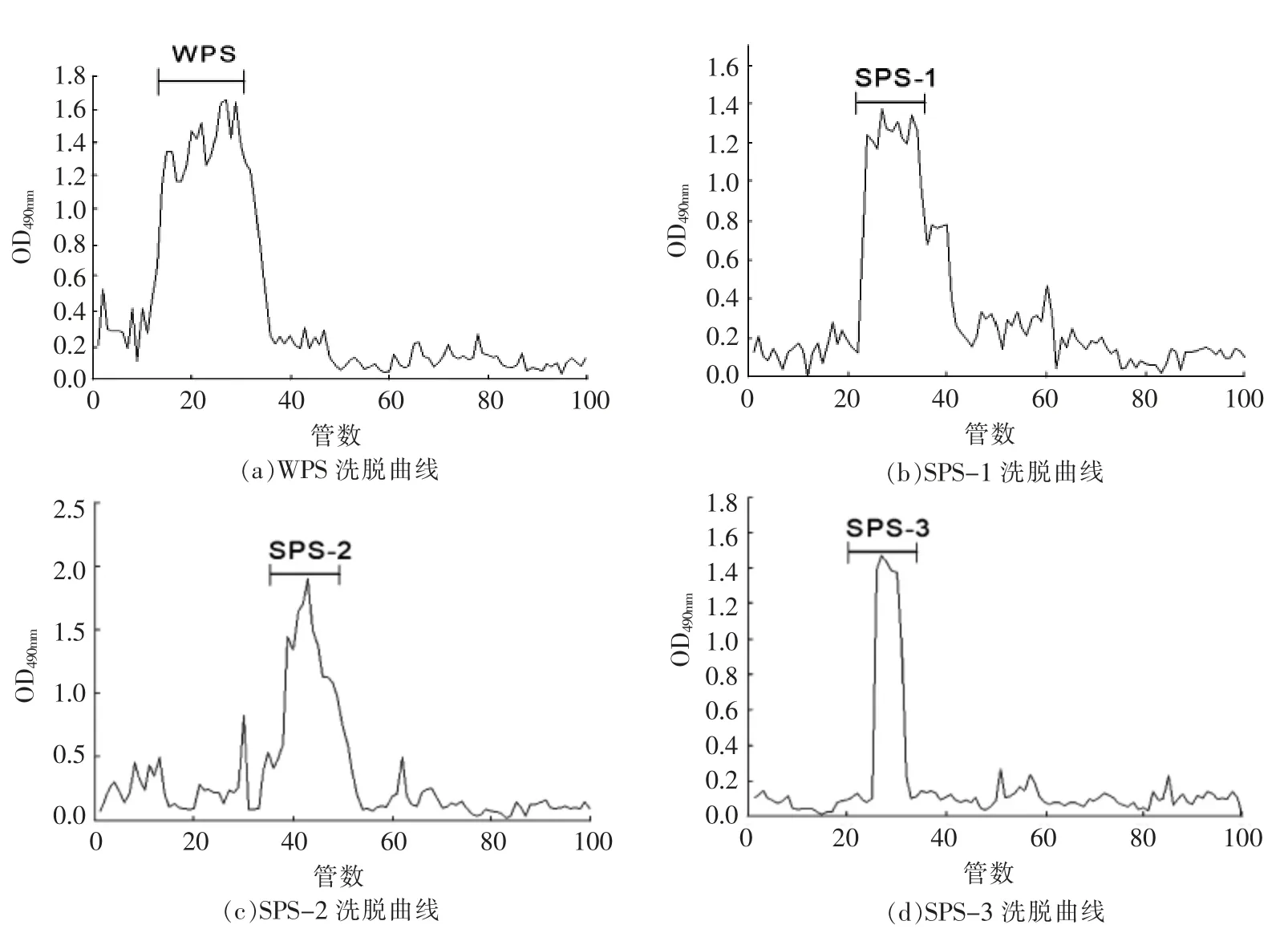
图4 Sepharose CL-6B 柱层析洗脱曲线Fig.4 Sepharose CL-6B column chromatography elution curve
2.2 GBRPs 结构解析结果
2.2.1 纯度及分子质量检测结果 GBRPs 各个多糖组分的UV-Vis 光谱大致相似仅选取一个为例,如图5。

图5 GBRPs 紫外-可见光光谱图Fig.5 The UV-Vis spectra of GBRPs
由紫外-可见光光谱分析可知,在波长260 nm 和280 nm 处均无明显吸收峰,表明各个组分纯度较好。采用GPC 检测其相对分子质量,线性回归方程为:lgMw=-0.1421tR+13.2451,式中:Mw为平均分子质量/ku;tR为保留时间/min;相关系数R2为0.9981。根据线性回归方程计算结果如下:WPS 的平均分子质量为1.47×105u,SPS-1、SPS-2以及SPS-3 的平均分子质量为9.62×105,5.59×106u 和3.15×105u。

图6 标准单糖和GBRPs 高效液相色谱图Fig.6 HPLC of the standard monosaccharides and GBRPs
2.2.2 单糖组成分析结果 高效液相色谱分析表明,WPS 组分的出峰时间为5.45,5.99,6.24,7.5,7.92 min,是由鼠李糖、木糖、阿拉伯糖、甘露糖和葡萄糖组成的,物质的量比为7.8∶9.1∶6.3∶3.9∶4.4;SPS-1 组分的出峰时间为5.5,7.59,7.69 min,是由鼠李糖、甘露糖和葡萄糖组成,物质的量比为8.3∶4.5∶10.1;SPS-2 组分的出峰时间为5.43,6.22,7.5,7.98 min,是由鼠李糖、阿拉伯糖、甘露糖和葡萄糖组成,物质的量比为10.3∶7.9∶6.7∶4.1;SPS-3 组分的出峰时间分别为5.38,6.19,7.94 min,是由鼠李糖、阿拉伯糖和葡萄糖组成,物质的量比4.8∶2.2∶6.5。
2.2.3 GBRPs 各组分官能团检测结果 GBRPs各组分在红外4 000~1 400 cm-1的扫描范围内都表现出了聚糖分子很明显的吸收峰,糖类分子中的O-H 伸缩振动峰在波数3 390 cm-1附近;2 700 cm-1附近的振动峰是C-H 伸缩振动峰;C=O 伸缩振动峰在波数1 645~1 690 cm-1附近;1 010 cm-1附近的振动峰是C-O-C、C-O-H 的伸缩振动峰;在波数1 200~800 cm-1范围内,多糖的特征峰表现出高度的专一性[25],其中WPS 在红外光谱1 200~1 700 cm-1之间有3 个明显的吸收峰,是吡喃糖环结构的吸收峰,表明WPS 中吡喃糖环的结构较多,WPS 在波数1 004.03 cm-1处出现明显的吸收峰,分析原因可能是醛基变角的振动,735.38 cm-1处有明显的吸收峰,原因可能是因为吡喃糖对称环状的振动,因此WPS 中可能含有D-吡喃糖环状结构[26]。SPS-2 组分在890.41 cm-1处的吸收峰是因为有β-糖苷键的存在,SPS-1、SPS-2 都含有β-糖苷键的结构。由吡喃环α 型C-H 键的变角振动所产生的吸收峰在830.33,844.61,828.99,841.37 cm-1处显示出来,说明α-糖苷键存在于WPS 中;而SPS-1 和SPS-2 的多糖组分中不但有α-糖苷键还有β-糖苷键结构;SPS-1 和SPS-3 在844.61 cm-1和841.37 cm-1处有吸收峰,分析原因可能是呋喃环中C1-H 的变角振动产生的吸收峰,因此这2 种多糖组分中可能含有α-D甘露吡喃糖[27];各GBRPs 组分在1 616 cm-1处没有明显的吸收峰,因为此范围内是氨基的特征吸收峰,所以说明GBRPs 在前期纯化的效果较好[28]。
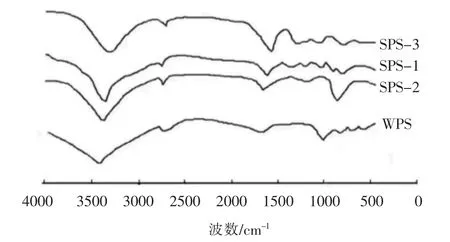
图7 GBRPs 的红外光谱图Fig.7 FTIR profiles of GBRPs
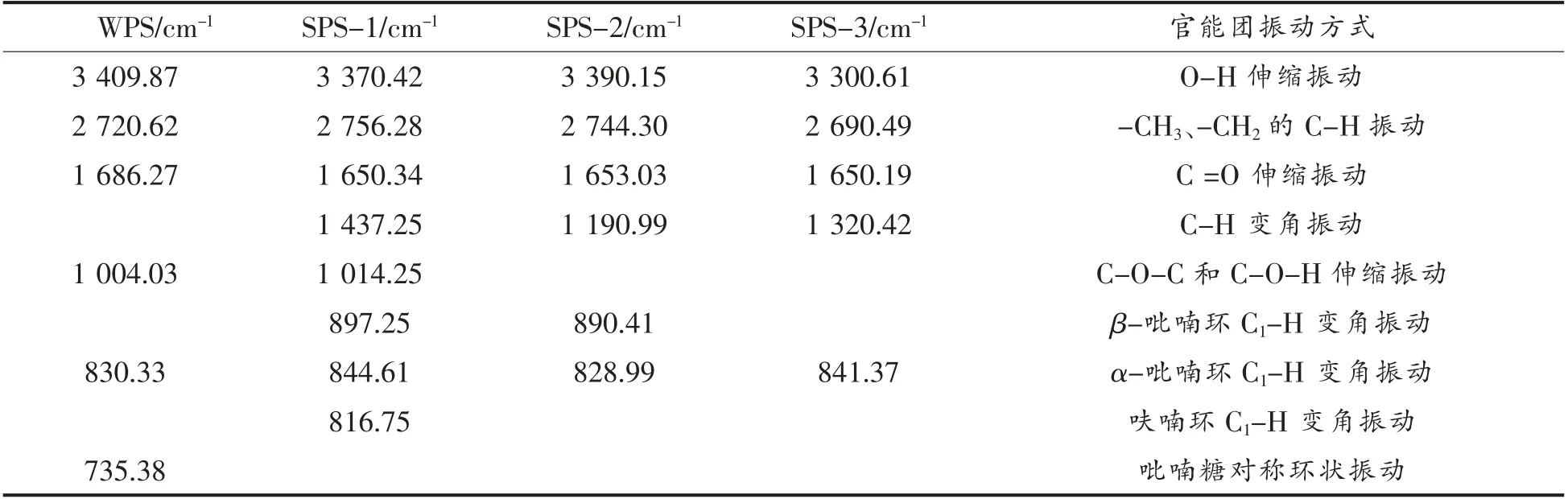
表1 GBRPs 各组分红外分析结果Table 1 The results of GBRPs FTIR analysis
2.2.4 GBRPs 各组分糖苷键构型检测结果 通过1H-NMR 光谱可以研究GBRPs 中糖苷键的构型。1H-NMR 中4.0~5.5 的区域是多糖的糖苷键中质子信号主要存在的区域,然而因为质子信号间的重叠与干扰的缘故,导致分析1H-NMR 谱图不是很清晰容易,所以要在4.8~5.5 这段区域内对首碳上的特征信号进行分析判断[29]。5.0 是区分吡喃糖构型的质子信号的临界值,首碳上的质子位移大于5.0 为α-型糖苷,小于5.0 为β-型糖苷。另外从氢谱中也可以判断某种均一多糖是否存在六元环吡喃糖还是五元环呋喃糖,如5.4 附近出现质子信号并且J1,2值小于2,则认为该物质属于呋喃糖[30]。从图8中可以看出,4.70 附近的谱峰是D2O中氘质子信号,WPS 主要存在α-型糖苷构型,结合红外的分析结果推测该处信号可能是α-Araf-(1→引起;SPS-1 则是α 和β-型糖苷构型。4.90,4.88 和4.85 可能是在β-Galp-(1→引起,且一维核磁氢谱结果与红外图谱结果具有一致性。
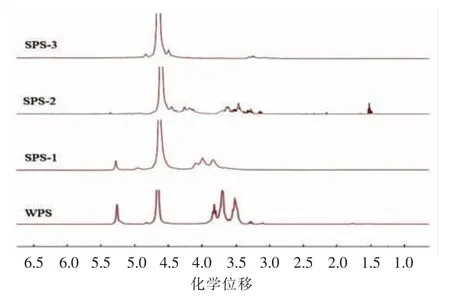
图8 GBRPs 核磁共振氢谱图Fig.8 1H-NMR spectra of GBRPs
2.3 GBRPs 的降血糖活性结果
2.3.1 GBRPs 不同组分对α-葡萄糖苷酶的抑制活性的影响 PNPG 是一种麦芽糖的类似物,能够被α-葡萄糖苷酶分解为对硝基苯酚,可通过检测对硝基苯酚的吸光度值计算样品中α-葡萄糖苷酶抑制剂的抑制率,如图9所示,经层析柱分离得到的GBRPs 4 个组分对α-葡萄糖苷酶均有一定程度的抑制作用,SPS-3 组分对α-葡萄糖苷酶抑制作用最小,抑制率为18.87%;SPS-1 组分对α-葡萄糖苷酶抑制抑制率最大,抑制率为57.36%,表明SPS-1 多糖组分中的酶抑制物质的含量较高。而SPS-3 多糖组分中酶抑制物质的含量最小;0.1 mol/L 的NaCl 溶液能够将大部分被吸附的酶抑制物质洗脱下来;WPS 组分和SPS-2 多糖组分中酶抑制物质含量相似,对α-葡萄糖苷酶抑制率基本相同。

图9 各组分多糖对α-葡萄糖苷酶的抑制作用Fig.9 Inhibitory effect of polysaccharides from each component on α-glucosidase
2.3.2 GBRPs 不同组分对胰岛素抵抗HepG2 细胞糖代谢的影响 HK 和PK 都是糖代谢过程中起到关键作用的2 种酶。在糖酵解和糖原合成的过程中己糖激酶是起到关键作用的限速酶。在糖酵解过程中丙酮酸激酶催化磷酸烯醇式丙酮酸生成丙酮酸,并将ADP 转化为ATP。在糖代谢过程中HK 和PK 在胰岛素抵抗细胞中都会减少[31]。从图中可以看出,胰岛素抵抗模型组与空白组相比,胰岛素抵抗模型组的HK 和PK 活力明显下降。加入SPS-1 多糖组分12 h 和24 h 后,IR-HepG2 细胞的PK 与HK 的活力明显提高,SPS-1 组分使糖的酵解过程加快,加快了细胞对葡萄糖的吸收。36 h 后,HK 和PK 酶活力开始下降,分析原因可能是长时间培养使一部分酶失活,使其酶活力下降。糖原的合成是胰岛素降低机体葡萄糖浓度的另外一条途径[32],由图12可知,模型组中IRHepG2 细胞糖原含量与对照组细胞相比下降了16.92%,加入SPS-1 后,IR-HepG2 细胞的糖原含量有明显的上升,可见SPS-1 对缓解胰岛素抵抗起到了明显的作用,提高了胰岛素抵抗细胞内的糖原含量。
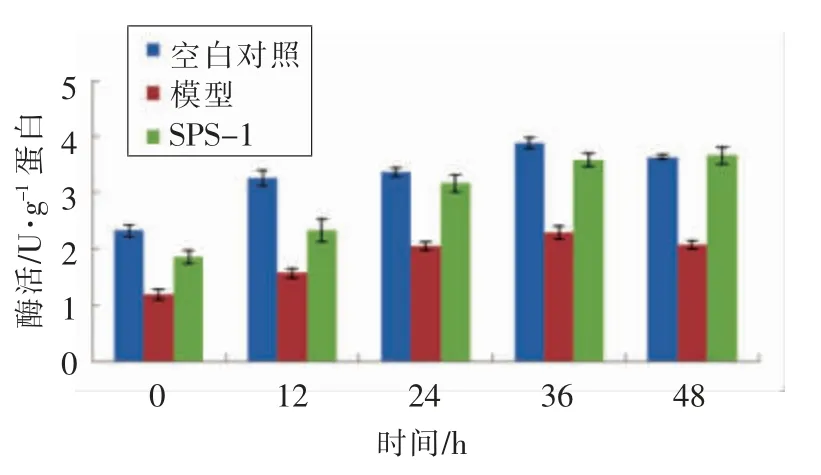
图12 SPS-1 对HepG2 细胞胰岛素抵抗细胞模型糖原合成的影响Fig.12 Effect of polysaccharides on glycogen synthesis in IR-HepG2 cell
3 结论
本试验通过水提醇沉法、酶-Savage 法以及大孔树脂吸附法提取并纯化发芽糙米多糖,GBRPs经脱色后的脱色率为86.57%,多糖损失率为28.96%。GBRPs 经两次酶-Sevage 法去除蛋白后,脱蛋白率为74.36%,多糖损失率为14.09%。采用离子交换柱与凝胶层析柱对GBRPs 进行分离,得到4 种多糖组分,分离效果较好。采用UV-Vis 检测各多糖组分纯度较好,GPC 分析检测了各组分多糖的分子质量,其中WPS 的平均分子质量为1.47×105u,SPS-1、SPS-2 以及SPS-3 的平均分子质量为9.62×105,5.59×106u 和3.15×105u。单糖分析表明,WPS 由鼠李糖、木糖、阿拉伯糖、甘露糖和葡萄糖组成,SPS-1 由甘露糖、鼠李糖和葡萄糖组成,SPS-2 由鼠李糖、阿拉伯糖、甘露糖和葡萄糖组成,SPS-3 由鼠李糖、阿拉伯糖、和葡萄糖组成。FTIR 分析显示,各多糖组分都含有多糖的特征吸收峰,WPS 主要由吡喃型糖环构成,SPS 中吡喃型与呋喃型糖环共存。1H NMR 分析表明,WPS 是含有吡喃环的α-构型,SPS 中存在α-和β-构型的呋喃和吡喃糖环。通过α-葡萄糖苷酶活性抑制及细胞试验,证实了SPS-1 具有α-葡萄糖苷酶活性抑制作用,且可以显著提高模型细胞HK 与PK 的活性,加快了糖酵解,使糖原含量升高,这为将来研究GBRPs 更深层次的功能作用机制提供理论参考价值。

图10 SPS-1 对HepG2 细胞胰岛素抵抗细胞模型PK 的影响Fig.10 Effect of polysaccharides on PK in IR-HepG2 cell
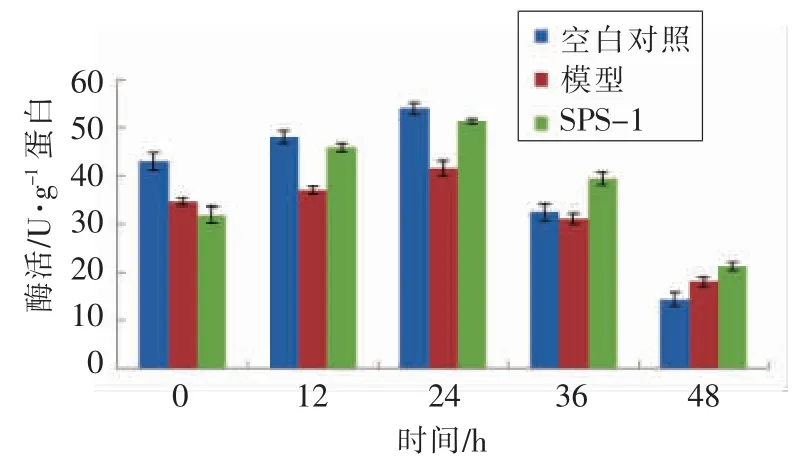
图11 SPS-1 对HepG2 细胞胰岛素抵抗细胞模型HK 的影响Fig.11 Effect of polysaccharides on HK in IR-HepG2 cell
