噻唑烯酮与芴硫叶立德[2+1]环加成反应合成双螺环化合物
2021-09-08王强万洪维顾承志杜广芬
王强,万洪维,顾承志,杜广芬
(石河子大学化学化工学院/新疆兵团化工绿色过程重点实验室,新疆 石河子 832003)
螺环作为化合物中一类重要的结构,在天然产物以及具有生物活性的化合物中广泛分布[1],具有螺环结构的化合物在抗菌[2]、糖尿病治疗[3],增强生物免疫效应[4]等方面有着显著成效。早期化学家会借助Simmons-Smith反应[5]得到环丙烷类化合物,通过进一步的反应可获得螺环骨架的化合物。随着化学家在合成方面的不断努力,近年来相继出现了多种多样的合成手段。2007年LIU L[6]等利用芳基二烯化物通过环化反应制备多取代的双环及多环螺环化合物,但反应过程需多步进行,不能一锅合成,而且反应过程还需添加叔丁基锂促进反应的进行,且由于叔丁基锂高度易燃而使反应操作的安全性能降低;GUO C[7]等利用不饱和醛经氮杂环卡宾催化制备螺环化合物,其[3+ 2]环化反应通过环化过程制备了一系列螺环化合物,但反应过程不仅需要催化剂的协助,而且反应的周期也较长,还需要在加热的条件下进行;FU F Y[8]等利用邻溴环丁酮在钯及膦催化剂的作用,并通过环化反应构建苯并螺环化合物的过程,以优异的产率制备了一系列苯并螺环化合物,但是该反应过程不仅需要多种催化剂的参与,还需要过渡金属的参与,使得反应过程不够经济,另外反应过程同样需要加热,而且温度高达100 ℃;YOU Y[9]等利用N-2,2,2-三氟乙基靛红酮亚胺分别与β-三氟甲基烯酮、3-三氟亚乙基苯并呋喃酮、3-三氟亚乙基吲哚通过[3+2]环加成反应构建3,2′-吡咯烷基螺氧辛酯的过程,该反应在构建结构多样的螺环化合物时表现出极高的效率,而且三氟甲基取代的吡咯烷部分还具有良好的立体控制性,但是该反应制备螺环化合物的过程条件较为苛刻,不仅需要手性催化剂的辅助,更需要在0 ℃甚至更低温的环境下进行。
本研究人员利用硫叶立德合成芳基-硫醚衍生物[10]以及用噻唑烯酮与苯炔进行反应,又在2019年利用噻唑烯酮与硫叶立德成功制备一系列噻唑类螺环产物[11],同年又利用芴衍生物与苯炔成功合成螺环吖啶化合物[12],在此基础上通过改变常规的硫叶立德,让芴取代的硫叶立德与活泼的噻唑烯酮反应去获得螺环产物,同时介于以上目前合成螺环化合物方法的现状,研发一种绿色高效合成螺环化合物的方法去改善现存方法存在的反应操作时安全性能低、需要过渡金属催化、反应条件苛刻等不足就显得非常必要。本文研究在合成螺环化合物时主要通过[2+1]环加成反应,以1.5当量Cs2CO3为反应的碱,无水甲苯为反应的溶剂,在常温下将1.5当量的芴硫叶立德与噻唑烯酮反应1 h即可获得目标产物。本文研究的意义在于首次利用芴硫叶立德与噻唑烯酮反应去制备双螺环化合物,而且在制备螺环化合物方面有以下独特的优势:首先实验无需过渡金属的催化,仅需要在无机碱Cs2CO3的作用下噻唑烯酮即可与硫叶立德进行反应;其次,反应在室温下就能进行,不需要加热以及低温等苛刻的反应环境,而且反应通过一锅法就能获得螺环化合物,不需要多步进行;最重要的是本反应的反应周期短,仅需要1 h就能完成螺环化合物的制备;另外,研究发现以噻唑环以及具有较大张力的环丙烷碳环为骨架的化合物在天然产物以及生物医药等领域有着重要的应用,其中以噻唑环为骨架的化合物在癌症治疗[13]、病毒防治[14]等方面有着良好的抗性,以环丙烷为骨架的化合物在抗真菌[15]领域有着不错的成效,本文研究所合成的化合物同时兼有噻唑环和环丙烷骨架,而且由文献[11]研究结果可知本文研究所合成的螺环化合物同样在病毒防治、抗菌、医药研发等领域有着不错的应用前景,本文对此反应相关的实验优化以及底物的适应性拓展的进行了讨论与总结。
1 实验部分
1.1 实验仪器与试剂
1H NMR (400 MHz),用TMS作为内标;采用Bruker Avance III HD 400 MHz型核磁共振仪,CDCl3为氘代试剂;所有溶剂严格按照溶剂处理手册标准进行处理;薄层色谱板为青岛海洋化工GF 254硅胶板;柱层析硅胶为青岛海洋化工厂生产(300~400目);硫叶立德前体[16]以及噻唑烯酮[17]的合成均参考相应的文献;其他化学试剂均为分析纯或化学纯,购买于阿拉丁化学试剂公司。
1.2 实验步骤和化合物的结构表征
1.2.1 噻唑烯酮与芴硫叶立德环加成合成双螺环产物及其衍生物的通用步骤
把25 mL的反应管放入红外干燥箱干燥30 min,氮气保护下冷却到室温,然后向反应管中加入准确称取的噻唑烯酮(0.2 mmol,54 mg)、硫叶立德前体(0.3 mmol,70 mg)、Cs2CO3(0.3 mmol,100 mg),最后再加入1.0 mL的无水甲苯溶剂,加料完成后在氮气保护和室温状态下搅拌反应,通过薄层色谱跟踪监测至原料消失。反应结束后将反应液转移到鸡心瓶中,向鸡心瓶中加入少量硅胶,借助旋转蒸发仪通过减压蒸馏除去溶剂并完成反应液固化,粗产物通过柱色谱分离纯化得到目标产物(硅胶200~300目,PE(石油醚)∶EA(乙酸乙酯)=10∶1作为洗脱剂)。
1.2.2 克级双螺环产物3a的制备方法步骤
将100 mL的史莱克瓶放入红外干燥箱干燥20 min,关闭红外干燥箱,冷却到室温后,然后向反应瓶中加入准确称取的芴系硫叶立德 2a(7.5 mmol,1.75 g)、Cs2CO3(7.5 mmol,2.5 g)和噻唑烯酮衍生物 1a(5 mmol,1.35 g),再加入25.0 ml无水甲苯,加料完成后在室温下搅拌反应,通过薄层色谱跟踪监测至原料消失。反应结束后将反应液转移到鸡心瓶中,向鸡心瓶中加入少量硅胶,借助旋转蒸发仪通过减压蒸馏除去溶剂并完成反应液固化,粗产物通过柱色谱析分离得到1.25 g 3a (硅胶200~300目,PE(石油醚)∶EA(乙酸乙酯)=10∶1作为洗脱剂),反应产率为80%。
1.2.3 目标化合物结构与表征
(3a)2′,3′-diphenyl 4″H-bisspiro [fluorene 9,1′-cyclopropane 2′,5″ -thiazole]4″-ketone:产率95%;1H NMR (400 MHz,CDCl3)δ8.22 (d,J=7.1 Hz,1H),8.03 (d,J=7.2 Hz,2H),7.78 (dt,J=7.6,1.3 Hz,2H),7.59 (t,J=7.5 Hz,1H),7.52-7.29(m,8H),7.26-7.23 (m,2H),7.02 (td,J=7.7,1.1 Hz,1H),6.16 (d,J=7.8 Hz,1H),4.65 (s,1H);13C NMR (100 MHz,CDCl3)δ191.4,187.6,142.0,140.7,140.2,140.0,134.7,132.4,132.1,130.3,128.9,128.4,128.4,128.3,128.2,127.7,127.1,126.7,125.4,125.4,120.0,119.6,57.0,51.9,41.1。HRMS (ESI) m/z 理论值 C29H20NOS+(M+H)+430.126 0,实测值 430.126 3。
(3b)3′-(4-fluorophenyl)-2″-phenyl-4″H-dipyrrole [fluorene 9,1′-cyclopropane 2′,5″-thiazole]-4″-ketone:产率83%;1H NMR (400 MHz,CDCl3)δ8.21(d,J=7.2 Hz,1H),8.03 (d,J=9.6 Hz,2H),7.79(d,J=7.6 Hz,2H),7.62-7.57 (m,1H),7.46-7.32(m,5H),7.24-7.19 (m,2H),7.07-7.00 (m,3H),6.16 (d,J=7.8 Hz,1H),4.58 (s,1H);13C NMR (100 MHz,CDCl3)δ191.3,187.5,163.8,161.3,142.0,140.7,140.1,139.7,134.8,132.1,132.0,129.0,128.5,128.4,127.8,127.2,126.6,125.5,125.4,120.1,119.7,115.6,115.3,56.9,51.8,40.3。HRMS (ESI) m/z 理论值C29H19FNOS+(M+H)+448.116 6,实测值448.116 5。
(3c)3′-(4-chlorophenyl) -2″-phenyl-4″H-bisspiro [fluorene 9,1′-cyclopropane 2′,5″ -thiazole]-4″-ketone:产率95%;1H NMR (400 MHz,CDCl3)δ8.20(d,J=7.9 Hz,1H),8.02 (d,J=9.5 Hz,2H),7.78(d,J=7.6 Hz,2H),7.60 (t,J=7.5 Hz,1H),7.55-7.27 (m,7H),7.18 (d,J=7.7 Hz,2H),7.06 (t,J=7.1 Hz,1H),6.18 (d,J=7.8 Hz,1H),4.57 (s,1H);13C NMR (100 MHz,CDCl3)δ191.4,187.4,142.0,140.7,140.0,139.6,134.9,134.3,132.0,131.7,130.9,129.0,128.7,128.5,128.4,127.9,127.2,126.5,125.6,125.4,120.1,119.7,56.7,51.6,40.4。HRMS (ESI) m/z 理论值C29H19ClNOS+(M+H)+464.087 0,实测值464.087 3。
(3 d)3′-(4-bromophenyl)-2″-phenyl-4″-H-dipyro[fluorene-9,1′-cyclopropane-2′,5″-thiazole]-4″-ketone:产率92%;1H NMR (400 MHz,CHCl3)δ8.20(d,J=7.9 Hz,1H),8.02 (dd,J=8.4,1.1 Hz,2H),7.78 (d,J=7.6 Hz,2H),7.60 (t,J=7.5 Hz,1H),7.48-7.33 (m,7H),7.12 (d,J=9.3 Hz,2H),7.07(t,J=7.7 Hz,1H),6.18 (d,J=7.8 Hz,1H),4.54 (s,1H);13C NMR (100 MHz,CDCl3)δ191.3,187.2,142.0,140.5.140.1,139.8,134.6,134.2,132.5,131.9,130.7,129.5,128.5,128.3,128.0,127.8,127.3,126.6,125.3,120.4,119.5,56.6,51.7,40.3。HRMS (ESI) m/z 理 论 值C29H19BrNOS+(M+H)+508.036 5,实测值508.036 7。
(3e)4′-oxo-2″-phenyl-4″H-dipyro[fluorene-9,1′-cyclopropane-2′,5″-thiazole]-3′-yl) benzo Nitrile:产率85%;1H NMR (400 MHz,CDCl3)δ8.20 (d,J=7.6 Hz,1H),8.01 (d,J=7.2 Hz,2H),7.79 (dd,J=7.2,2.7 Hz,2H),7.64 (d,J=8.3 Hz,3H),7.47-7.33 (m,8H),7.05 (t,J=7.2 Hz,1H),6.06 (d,J=7.8 Hz,1H),4.61 (s,1H);13C NMR (100 MHz,CDCl3)δ191.2,187.1,142.1,140.7,139.7,139.2,137.8,135.1,132.2,131.8,131.2,129.0,128.6,128.5,128.1,127.4,126.1,125.7,125.4,120.3,119.8,118.4,112.3,56.3,51.3,40.4。 HRMS(ESI) m/z 理论值C30H19N2OS+(M+H)+455.121 3,实测值455.121 7。
(3f)2′-phenyl-3′-(p-tolyl)-4″H-dipyro [fluorene-9,1′-cyclopropane-2′,5″-thiazole]-4″-ketone:产 率96%;1H NMR (400 MHz,CDCl3)δ8.21 (d,J=8.1 Hz,1H),8.02 (d,J=8.5 Hz,2H),7.78 (d,J=7.5 Hz,2H),7.58 (t,J=7.5 Hz,1H),7.46-7.37 (m,4H),7.33(t,J=7.5 Hz,1H),7.13 (d,J=1.3 Hz,4H),7.03(t,J=7.6 Hz,1H),6.22 (d,J=7.8 Hz,1H),4.60(s,1H),2.21 (s,3H);13C NMR (100 MHz,CDCl3)δ191.4,187.7,142.0,140.7,140.3,140.1,138.0,134.7,132.2,130.2,129.3,129.1,128.9,128.4,128.2,127.6,127.1,126.8,125.4,119.9,119.6,57.1,52.0,41.0,21.3。HRMS (ESI) m/z 理论值C30H22NOS+(M+H)+444.141 7,实测值444.141 8。
(3 g)3′-(4-methoxyphenyl)-2″-phenyl-4″H-dipyro [fluorene-9,1′-cyclopropane-2′,5″-thiazole]-4″-ketone:产率92%;1H NMR (400 MHz,CDCl3)δ8.21(d,J=7.2 Hz,1H),8.02 (d,J=9.6 Hz,2H),7.78(d,J=7.5 Hz,2H),7.61-7.55 (m,1H),7.46-7.39 (m,4H),7.33 (t,J=7.1 Hz,1H),7.15 (d,J=8.8 Hz,2H),7.04 (t,J=7.1 Hz,1H),6.85 (d,J=8.8 Hz,2H),6.23 (d,J=7.8 Hz,1H),4.58 (s,1H),3.82 (s,3H);13C NMR (100 MHz,CDCl3)δ190.4,187.6,141.8,140.0,139.6,136.8,132.1,130.9,130.0,129.3,129.1,128.6,128.0,127.5,126.6,125.5,121.2,113.8,57.3,55.7,52.1,41.0。HRMS (ESI) m/z 理 论 值C30H22NO2S+(M+H)+460.136 6,实测值460.136 3。
(3 h)3′-(2-chlorophenyl)-2″-phenyl-4″H-bisspiro[fluorene9,1′-cyclopropane 2′,5″-thiazole]-4″-ketone:产率41%;1HNMR (400 MHz,CDCl3)δ8.26-8.22 (m,1H),8.00-7.94 (m,2H),7.82-7.76(m,2H),7.50-7.20(m,10H),7.10-7.00 (m,1H),6.31 (d,J=7.90 Hz,1H),4.50(s,1H);13C NMR(100 MHz,CDCl3)δ190.1,187.2,141.9,141.2,140.7,140.3,139.9,136.5,131.4,130.6,130.0,129.6,129.3,128.4,127.9,127.3,126.3,125.4,120.1,119.6,57.2,51.8,40.9。
(3i)3′-(3-methoxyphenyl) -2″-phenyl-4″H-dipyro [fluorene-9,1′-cyclopropane-2′,5″-thiazole]-4″-ketone:产 率80%;1H NMR (400 MHz,CDCl3)δ8.25 (d,J=8.3 Hz,1H),7.97 (d,J=8.7 Hz,2H),7.80-7.77 (m,2H),7.48-7.26 (m,11H),7.05(t,J=7.6 Hz,1H),6.31 (d,J=7.8 Hz,1H),4.50(s,1H),3.71 (s,3H);13C NMR (100 MHz,CDCl3)δ190.1,187.2,141.9,141.2,140.7,140.2,139.9,136.6,131.4,130.6,130.0,129.7,129.3,128.4,127.9,127.3,126.3,125.6,120.1,119.7,57.2,55.4,51.8,40.9。
(3j)3′-([1,1′-biphenyl]-4-yl) -2′-phenyl-4″Hdipyro [fluorene-9,1′-cyclopropane-2′,5″-Thiazole]-4′-ketone:产率87%;1H NMR (400 MHz,CDCl3)δ8.24(d,J=7.2 Hz,1H),8.04 (d,J=9.7 Hz,2H),7.80(d,J=7.5 Hz,2H),7.64-7.56 (m,5H),7.48-7.30(m,11H),7.07-7.02 (m,1H),6.28 (d,J=7.8 Hz,1H),4.67 (s,1H);13C NMR (100 MHz,CDCl3)δ191.5,187.2,141.3,140.3,139.9,134.7,132.1,131.4,130.7,128.9,128.4,127.7,127.2,127.0,126.7,125.5,125.4,119.9,119.6,57.3,52.1,40.8。
(3k)3′-(furan-2-yl)-2″-phenyl-4″H-dipyroro[fluorene-9,1′-cyclopropane-2′,5″-thiazole]-4″-ketone:产率95%;1H NMR (400 MHz,CDCl3)δ8.08(d,J=8.1 Hz,1H),7.95 (d,J=9.6 Hz,2H),7.68(d,J=7.8 Hz,2H),7.51 (t,J=7.5 Hz,1H),7.41-7.24 (m,6H),7.05 (t,J=8.2 Hz,1H),6.46-6.40(m,2H),6.35 (dd,J=3.3,1.9 Hz,1H),4.35 (s,1H);13C NMR (100 MHz,CDCl3)δ190.2,172.5,154.4,145.2,143.3,141.7,138.7,134.8,134.6,134.0,129.4,129.1,127.8,127.5,127.0,125.8,125.2,121.2,120.3,119.5,51.5,36.7,35.6。
(3l)3′-(thien-2-yl)-2″-phenyl-4″H-dipyrrole[fluorene-9,1′-cyclopropane-2′,5″-thiazole]-4″-ketone:产率97%;1H NMR (400 MHz,CDCl3)δ8.36(d,J=7.4 Hz,1H),7.68-7.63 (m,3H),7.55-7.45 (m,6H),7.30-7.25 (m,6H),3.95(s,1H);13C NMR (100 MHz,CDCl3)δ193.8,171.3,154.0,144.5,143.0,141.3,141.4,138.2,134.7,129.2,129.1,127.5,126.8,125.7,125.0,124.3,120.3,119.9,118.8,51.3,35.9,35.4。
(3 m)3′-phenyl-2″-(4-chlorophenyl)-4″H-bisspiro[fluorene9,1′-cyclopropane 2′,5″-thiazole]-4″-ketone:产率85%;1HNMR (400 MHz,CDCl3) δ 8.21(d,J=8.0 Hz,1H),8.17-8.13 (m,1H),7.97-7.90 (m,2H),7.80-7.75 (m,2H),7.68-7.63(m,1H),7.55-7.50 (m,2H),7.42-7.35 (m,2H),7.35-7.30 (m,2H),7.24-7.21 (m,2H),7.01(t,J=7.6 Hz,1H),6.15 (d,J=7.8 Hz,1H),4.64 (s,1H);13C NMR (100 MHz,CDCl3) δ 189.9,187.3,185.9,182.9,142.0,141.7,140.6,139.9,139.0,133.7,132.2,131.2,129.6,128.4,128.3,127.8,126.3,125.3,120.0,57.3,52.0,41.2。
(3n)3′-(4-methoxyphenyl)-2″-(4-chlorophenyl)-4″H-dipyroro[fluorene-9,1′-cyclopropane-2′,5″-Thiazole]-4″-ketone:产 率 82%;1HNMR (400 MHz,CDCl3) δ 8.20 (d,J=6.4 Hz,1H),8.05-7.98 (m,2H),7.80-7.74 (m,2H),7.62-7.52(m,2H),7.48-7.30 (m,5H),7.18-7.10 (m,2H),7.08-7.00(m,1H),6.90-6.80 (m,2H),6.23 (d,J=8.0 Hz,1H),4.58 (s,1H),3.81 (s,3H);13C NMR (100 MHz,CDCl3) δ 191.3,187.7,159.4,141.9,140.6,140.3,140.1,134.7,132.1,131.5,128.9,128.2,127.6,126.8,125.4,124.3,119.9,119.6,113.7,57.2,55.3,52.0,40.6。
(3o)3′-(4-methoxyphenyl) -2″-(4-methoxyphenyl)-4″H-dipyroro [fluorene-9,1′-cyclopropane-2′,5″-Thiazole]-4′-ketone:产率78%;1H NMR (400 MHz,CDCl3) δ 8.25-8.18 (m,1H),7.97-7.91 (m,2H),7.81-7.74 (m,2H),7.46-7.29 (m,3H),7.17-7.11 (m,2H),7.02 (td,J=7.6,1.2 Hz,1H),6.88-6.83 (m,4H),6.22 (d,J=7.8 Hz,1H),4.56 (s,1H),3.82 (s,3H),3.81 (s,3H);13C NMR (100 MHz,CDCl3) δ 189.98,187.65,165.07,159.34,140.22,131.53,130.72,128.08,127.53,127.07,126.84,125.49,125.38,124.80,124.51,119.87,119.54,114.24,113.70,57.26,55.63,55.30,51.65,40.30。
2 结果与讨论
2.1 噻唑烯酮与芴硫叶立德环加成反应
其反应式如下:

2.2 噻唑烯酮与芴硫叶立德环加成合成双螺环的条件优化
以噻唑烯酮与芴硫叶立德作为模型反应,对反应条件进行优化,见表1。

表1 反应条件优化
首先在无水甲苯中,以Cs2CO3为模型反应的碱,室温下反应。借助TLC监测反应至原料消失,反应进行了1 h,最后通过柱色谱分离纯化以95%的产率得到目标产物(表1序号1)。在此基础上进一步对其他常用的无机碱和有机碱进行筛选,但当用无机碱Na2CO3和K2CO3做反应的碱时,通过TLC监测发现,反应进行1 h后并没有以Cs2CO3为反应的碱时的反应彻底,而是原料仍有大量剩余,于是延长反应时间至反应原料消失,通过柱色谱分离纯化分别以80%和85%的产率获得目标产物,且dr比均在20∶1以上(表1序号2、3);而当用有机碱Et3N、DIPEA、DBU、DMAP、DABCO、Imidazole作反应的碱时,在反应发生1 h后,通过TLC监测发现原料同样有大量剩余,选择继续延长反应时间并同时进行有效的反应监测,结果在反应进行24 h后,监测发现反应进行地很微弱,反应液中仍然存在大量的原料,最后将粗产物通过柱色谱分离发现,这些有机碱用作反应的碱时,产率均不足10%(表1中序号4~9)。
接着以Cs2CO3为反应的碱对反应溶剂进行筛选,发现大部分所筛选的溶剂是可以以优异的产率得到目标产物,其中以THF、DCM、CH3CN作溶剂、反应进行1 h后,发现原料消失,通过柱色谱的分离纯化,分别以92%、93%、90%的优异产率获得了目标产物(表1序号10~12),但是用DMF作反应的溶剂时,反应1 h后,原料并没有消失完,同样选择继续延长反应的时间,而且随着反应时间的延长,反应原料在慢慢减少,直至反应进行24 h后,原料消失,经柱色谱分离纯化,最终以80%的产率获得目标产物(表1序号13),尽管用THF、DCM、CH3CN以及DMF做反应溶剂以优异的产率获得了目标产物,但是产物的dr比却在20∶1以下,甚至不足10∶1 (表1序号10~13);同时反应即使以Cs2CO3为反应的碱,甚至延长反应的时间,有些反应仍然很微弱,如以DMSO和H2O作溶剂时,反应产率均在10%以下(表1中序号14、15);最后,以无水甲苯为溶剂,以Cs2CO3为反应的碱,保持其他条件不变的情况下,降低了Cs2CO3的加入量,即使延长反应的时间,反应产率也没有达到预期优异的产率,仅以81%的产率得到目标产物(表1序号16)。
因此,确定的最优反应条件为:以1.5当量的Cs2CO3为反应的碱,以无水甲苯为反应的溶剂,在常温下让1.5当量的芴硫叶立德与1a反应1 h即可。
2.3 噻唑烯酮与芴硫叶立德环加成合成双螺环的底物拓展
通过对上述条件的优化,本文确定了反应的最优条件,在最优的反应条件下对不同的噻唑烯酮和芴硫叶立德底物进行了底物的适应性研究。
由表2可知:
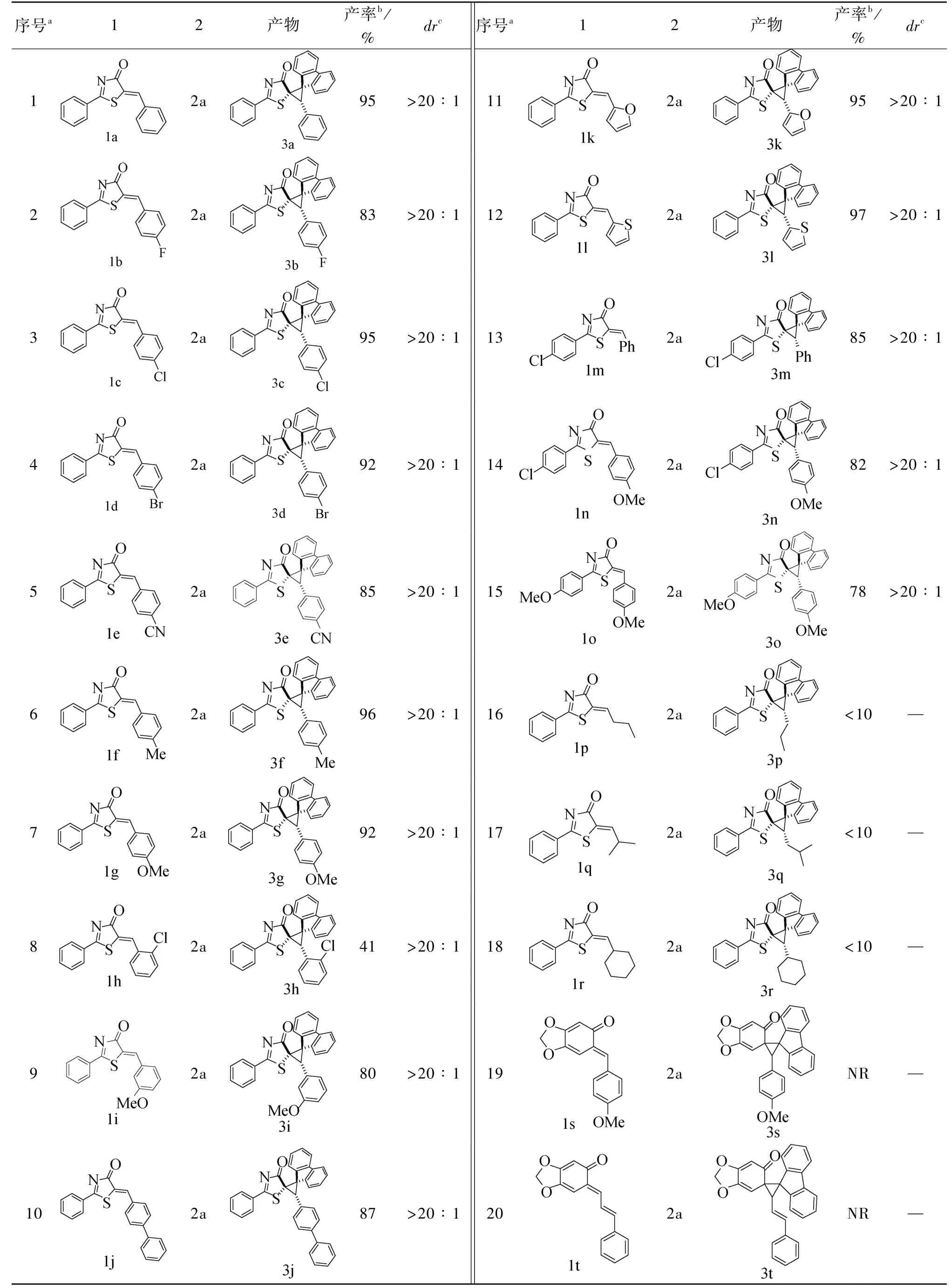
表2 反应底物普适性研究
(1)在研究噻唑烯酮芳环上电性不同的取代基对反应产率的影响时发现:吸电子取代基(Ar1=4-FC6H4,4-ClC6H4,4-BrC6H4,4-CNC6H4)和供电子取代基(Ar1=4-MeC6H4,4-MeOC6H4)取代的噻唑烯酮参与的环加成反应,都以较高的产率生成相应的双螺环的加成产物(表2序号1~7),这表明噻唑烯酮芳环上取代基的电子性质对反应产率的影响很小。
在研究噻唑烯酮芳环上吸电子以及给电子取代基的位置对反应产率的影响时,通过表2中具有代表性基团发现:噻唑烯酮芳环上给电子基的位置改变时(Ar1=3-MeOC6H4),对反应的影响很小,仍以80%的产率获得目标产物(表2序号9),但芳环上吸电子基的位置由对位变为邻位时(Ar1=2-ClC6H4),产率降低很多,仅有41%(表2序号8)。这可能与邻位导致空间位阻的增大有关,即噻唑烯酮与硫叶立德加成成环的阻碍增大,使得反应的产率降低。
当噻唑烯酮的芳环的对位连有苯基时(Ar1=4-Biphenyl),同样以87%优异的产率得到了相应的产物3j(表2序号10);此外,当取代基是杂环基团时(Ar1=2-furanyl,2-thienyl),仍分别以95%和97%较高的产率得到了目标产物3k-3l(表2序号11、12)。
(2)对噻唑烯酮芳环 Ar2连有不同取代基的研究发现:无论是吸电子基还是给电子基,都以优异的产率获得了目标产物3m-3o(表2中序号13~15)。
(3)用连有脂肪烃的噻唑烯酮与硫叶立德反应时(表2序号16~18)发现:噻唑烯酮所连有的脂肪烃不论是链状的还是环状的,反应均进行的复杂而且微弱,这可能是这类型的噻唑烯酮进攻硫叶立德加成成环后形成的加成产物自身不够稳定而容易发生分解导致的。
最后用芝麻酚衍生的醌酮与硫叶立德进行环加成反应,但反应并没有发生,可能是由于芝麻酚衍生的醌酮的电子云密度较大,使得此类醌酮化合物的亲电性减弱,从而无法使反应进行(表2产物19、20)。
(4)噻唑烯酮与芴硫叶立德反应所得的产物,除了具有优异的产率外,还有较好的dr比,而且通过对产物3a结构进行单晶衍射分析,由此确定了产物的非对映异构体的相对构型。
3a的单晶结构如图1所示。
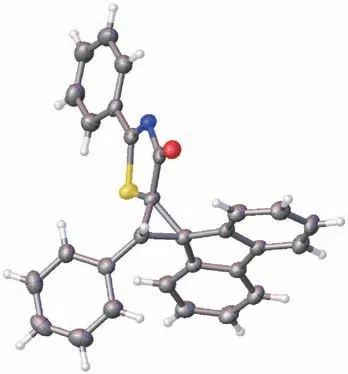
图1 3a的X射线晶体结构
3a单晶结构数据如下:分子式C29H19NOS;分子量43.66 g/mol;温度296(2) K;波长0.710 73 Å;单晶体系为orthorhombic;空间基团P b c a,a=18.790(5)Å(α=90°),b=8.100 1(18)Å(β=90°),c=28.102(6)Å(γ=90°);体积4 277.1(17) Å3;Z值为51;密度0.864 g/cm3;吸光系数t为0.089 mm-1;F000为1 138;数据收集n为1.45°~25.94°;索引范围-23≤h≤23,-9≤k≤9,-32≤l≤34;反射收集32 239;独立映像为4 154 [Rint=0.121 9];细化方法为Full-matrix least-squares onF2;细化参数为SHELXL-2018/3 (Sheldrick,2018);函数最小化Σw(Fo2-;数据/限制/参数为4154/0/290;拟合优度F2为0.998;吻合指数R为2658 data;I>2σ(I),R1=0.051 9,wR2=0.128 9,all data,R1=0.090 8,wR2=0.151 8;加权为w=1/[σ2(Fo2)+(0.075 1P)2],P=(Fo2+2Fc2)/3;消光系数0.002 1(4);最大衍射峰和孔为0.310、-0.249 eÅ-3;R.M.S.均值偏差为0.064 eÅ-3。
2.4 机理分析
基于在噻唑烯酮与硫叶立德环加成[11]方面的研究工作,再结合对本研究实验工作的分析总结,提出了其合理的机理,如图2所示。
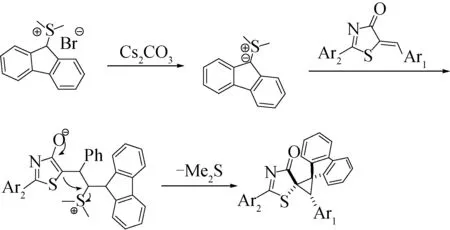
图2 反应机理
首先,芴硫叶立德前体在Cs2CO3的作用下生成稳定的芴硫叶立德,然后稳定的芴硫叶立德与噻唑烯酮经过Michael共轭加成得到中间体,该中间体通过进一步的分子内亲核进攻以及二甲基硫醚的离去最终得到目标产物。
3 结论
(1)本文研究在Cs2CO3作用下首次利用噻唑烯酮与芴硫叶立德通过[2+1]环加成反应得到双螺环化合物的过程,通过该反应过程实现了一种新的C—C键构建方法,与以往方法相比,这种新方法无需金属催化,产率高,获得的目标产物的选择性好,原子经济,反应条件温和、高效,底物适用性好,反应易于放大。
(2)噻唑烯酮与芴硫叶立德环加成反应的最佳条件:以1.0 ml无水甲苯为溶剂,0.2 mmol噻唑烯酮,0.3 mmol芴硫叶立德,在0.3 mmol 无机碱Cs2CO3的作用下,室温下反应1 h,以41%~97%的产率制备了15种目标产物。
(3)不论噻唑烯酮的烯烃位连有的是吸电子基还是给电子基的芳环,还是噻唑烯酮的烯烃位连有的是杂环,都可以很好地参与到环加成反应中,并以中等至优异的产率获得目标产物,这很好地拓展了噻唑烯酮与芴硫叶立德的环加成反应。
