β淀粉样蛋白免疫治疗阿尔茨海默病的研究进展
2021-09-07张梦泽管阳太
张梦泽,王 侃,管阳太
上海交通大学医学院附属仁济医院神经内科,上海 200127
阿尔茨海默病(Alzheimer’s disease,AD)是一种起病隐匿、进展缓慢的神经退行性疾病。AD 的病程根据认知能力和身体机能的恶化程度分成4 个时期:临床前期、临床早期、临床中期和临床晚期[1]。临床前期介于认知能力正常与失忆症之间,属于轻度认知障碍(mild cognitive impairment,MCI)。临床早期最常见的症状为短期记忆丧失,当疾病逐渐发展,可能出现语言障碍、定向障碍、情绪不稳、生活无法自理和行为问题。
2015 年,全球约有2 980 万人罹患AD,发病年龄一般为65 岁以上[2]。中国的AD 患病率和发病率较高,2019 年开展的大样本多区域研究的结果显示,>65 岁人群的痴呆患病率为5.60%,并且农村人口的痴呆患病率显著高于城市人口[3]。AD 给患者及其家属带来沉重的经济负担,并且严重影响老年人的生活质量。
目前,临床上用于治疗AD 的药物无法从根本上阻止疾病的进程,因此研发新的AD 治疗药物刻不容缓。2021 年,美国食品药品监督管理局(Food and Drug Administration,FDA)批准了Biogen 公司开发的aducanumab 治疗早期AD 的生物制品许可,成为2003 年以来美国FDA 批准的首个治疗AD 的新药,开创了AD 免疫治疗的新时代。当前的AD 免疫治疗临床试验主要围绕靶向β 淀粉样蛋白(amyloid β-protein,Aβ)和Tau 蛋白开展研究。本文介绍了靶向Aβ 的免疫治疗的研究现状。
1 AD 的病理学机制
AD 的病理学机制极为复杂,其典型的病理学特征是淀粉样斑块和神经原纤维缠结(neurofibrillary tangles,NFTs)。
淀粉样斑块位于神经元外,以Aβ 为中心,周围还包括各种蛋白聚糖、糖胺聚糖、核酸、金属离子和脂质等物质[4]。遗传学和生物化学分析的结果显示,Aβ 沉积介导了AD 的病理过程;而Tau 蛋白的聚集则与大脑的神经变性有关[5]。Aβ 由大脑中正常的大分子肽类物质淀粉样蛋白前体(amyloid precursor protein,APP)衍生而来,含39~43 个氨基酸残基。APP 由21 号染色体编码,是一种跨膜蛋白,对神经元的生长、存活和修复均至关重要。APP 由α、β 和γ 3 种分泌酶参与代谢,经β 分泌酶和γ 分泌酶的顺次切割后生成Aβ,并释放Aβ 至细胞外。经此切割产生的Aβ C 端的长度存在差异,每一种Aβ系根据其氨基酸残基数予以命名。Aβ40 是生理条件下的主要产物,而Aβ42 和Aβ43 则呈现出高度聚集倾向和神经毒性[6]。
目前尚不明确AD 的具体病理机制,但是大量的实验数据支持淀粉样蛋白级联假说[7]。该假说认为,Aβ 在脑实质中的缓慢沉积是AD 发展的关键步骤,细胞外的Aβ 单体逐渐聚集形成寡聚物、原纤维和纤维,直至形成不溶性斑块,出现淀粉样斑块和NFTs,最终导致神经元变性死亡(图1)。研究认为,Aβ 的突触毒性和神经毒性是由聚集过程中的可溶性寡聚物介导的[7]。正常状态下,中枢神经系统中的Aβ 可以通过多种方式得以清除,包括吞噬作用、免疫反应和蛋白水解降解;但是,在家族性和散发性AD 患者的大脑中,Aβ 的过量产生和(或)清除减少,可导致Aβ 的表达水平失衡。细胞外的Aβ 聚集可引发突触功能障碍和神经元毒性,还可以通过诱导氧化应激反应和神经炎症,导致神经元死亡。
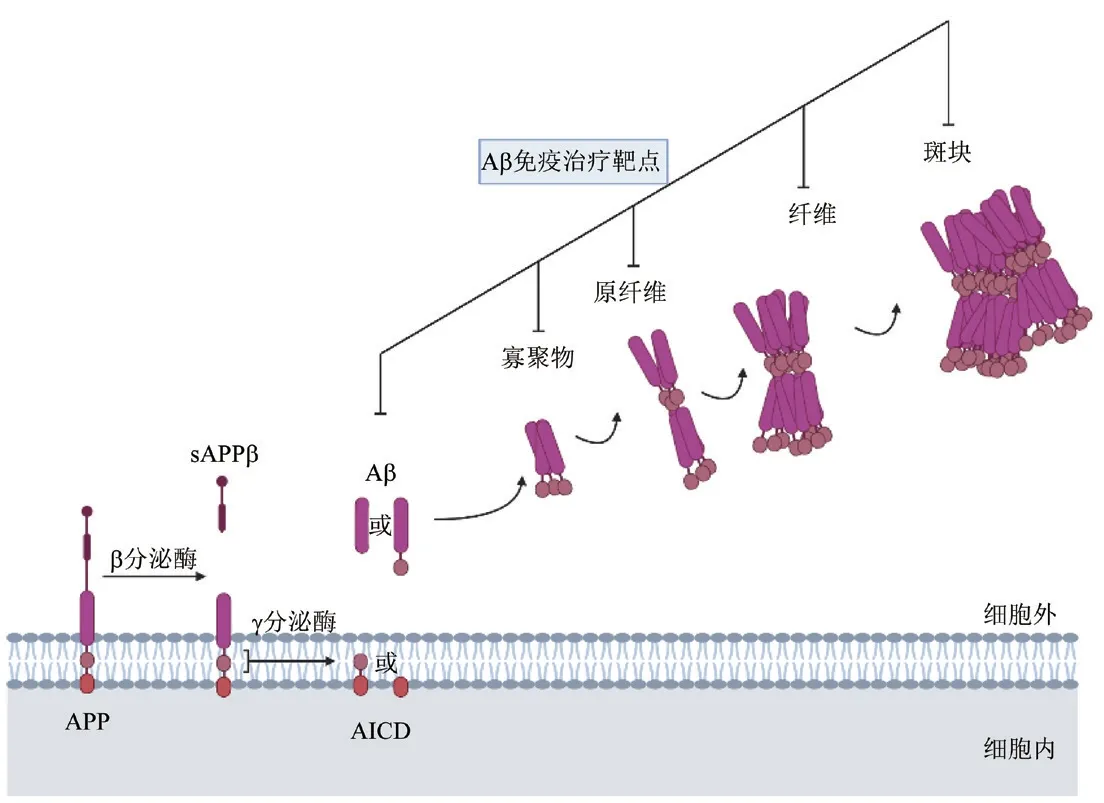
图1 Aβ 产生的病理学机制。Aβ:β 淀粉样蛋白;APP:淀粉样蛋白前体;sAPPβ:可溶性β 淀粉样蛋白前体;AlCD:淀粉样蛋白前体胞内结构。
NFTs 位于神经元细胞质内,主要由过度磷酸化的Tau 蛋白集合而成。Tau 蛋白属于微管相关蛋白,负责调控微管的稳定性[8]。病理状态下,过度磷酸化的Tau 蛋白从微管中分离,从而破坏了神经元微管的稳定性,并且在细胞质内和树突中异常聚集,形成NFTs,导致突触功能障碍和神经元死亡[9]。
2 靶向Aβ 的免疫治疗
靶向Aβ 的免疫治疗主要基于AD 患者的Aβ沉积于神经元细胞外的病理学特征,目的是通过特异性抗Aβ 抗体,抑制Aβ 聚集或促进其清除,进而发挥阻止其所致的神经变性的作用[10-11]。抗Aβ 抗体通过与Aβ 单体、寡聚物和原纤维相互作用,可以从多个水平干扰淀粉样蛋白级联反应。目前已发展出多种靶向Aβ 的免疫治疗策略。
根据抗体的来源不同,可以分为主动免疫和被动免疫。从传染病学的角度来看,主动免疫主要应用于感染发生之前传染病的预防,被动免疫则应用于感染发生之后传染病的治疗。对AD 的治疗也可以采取类似的策略,在AD 进展的不同阶段,采取不同的免疫治疗策略,从而达到预防和治疗AD 的效果。
2.1 Aβ 主动免疫治疗
Aβ 主动免疫治疗是通过注射Aβ 或其片段,诱导人体的免疫系统产生针对Aβ 的抗体。与被动免疫治疗相比,主动免疫治疗可以诱导出更持续的针对多克隆Aβ 的特异性免疫反应[12]。早在1999年,SCHENK 等[13]给予转基因AD 小鼠模型接种Aβ42 疫苗,结果显示可以抑制低龄小鼠Aβ 斑块的形成,缓解高龄小鼠AD 样神经病理的进展。
2.1.1 AN1792 疫苗
AN1792 疫苗是首个进入临床试验的抗Aβ 疫苗,由Aβ42 免疫原和QS21 佐剂组成,通过肌内注射给药。然而,由于6%(18/298)的AD 患者在治疗中出现无菌性脑膜炎,因此入组轻至中度AD 患者的Ⅱ期临床试验被迫于2002 年终止[14]。这种严重的不良反应被认为是由于AN1792 疫苗中的Aβ42 中心含有活化T 细胞的T 细胞表位,因此接种后引起T 细胞和小胶质细胞的活化[15]。即便如此,AN1792 疫苗针对Aβ 的功效获得了验证:与未产生抗体的AD 患者相比,产生抗体应答的AD 患者脑实质内的Aβ 斑块得以清除,并且其清除程度与免疫反应程度相关,而且在为期1 年的观察期间,AD 患者保持认知能力的稳定[16]。随访研究的结果显示,接种AN1792 疫苗4.6 年后,有抗体应答的AD 患者可维持很低水平的抗体滴度,并且其日常活动受损较安慰剂组有所减少[16]。尸检结果表明,AD 患者在接种AN1792 疫苗14 年后,大脑中仍可保持无Aβ 斑块[17]。
AN1792 疫苗的临床试验结果证实了Aβ 主动免疫治疗的可行性。为避免发生脑膜炎等不良反应,第2 代抗Aβ 疫苗不含T 细胞表位,其将免疫反应导向Aβ N 端的B 细胞表位[18]。
2.1.2 阿米莫肽(amilomotide,CAD106)疫苗
CAD106 疫苗是第2 代抗Aβ 疫苗,由QB 噬菌体包裹多重拷贝的Aβ N 端(Aβ1~6)小片段所构成,旨在诱导产生针对Aβ N 端的特异性抗体,同时不引起Aβ 特异性T 细胞反应[19]。Ⅰ期和Ⅱ期临床试验已经证明,CAD106 疫苗对轻至中度AD 患者是安全且可耐受的,能够产生令人满意的抗体反应,并且长期治疗可以延缓抗体滴度的降低[19-20]。CAD106 疫苗的主要不良反应是头痛和注射部位反应。一项Ⅱb 期临床试验的结果显示,CAD106 疫苗(450 μg)+明矾佐剂治疗轻度AD 可维持疗效与耐受性之间的平衡[21]。由Novartis/Amgen 公司合作开展的Ⅱ~Ⅲ期试验从2015 年开始招募载脂蛋白E 等位(apolipoprotein E allele,APOE)ε4 基因纯合的无认知障碍的临床前期AD 患者,旨在评估CAD106 疫苗对认知和潜在AD 临床发作风险的影响。该试验已于2020年7 月终止,目前还未发布任何结果。
2.1.3 ABvac40 疫苗
ABvac40 疫苗(Araclon Biotech 公司研制)由与锁孔帽贝血蓝蛋白(keyhole limpet hemocyanin,KLH)偶联的Aβ40 的B 细胞表位Aβ33~40 肽段构成,是最早靶向Aβ40 肽段C端的疫苗之一[22]。与靶向Aβ N 端的抗体不同,ABvac40 疫苗产生的抗体不会识别插入细胞膜的Aβ 前体APP,避免了潜在的神经元毒性,并且可以识别N 端截短或修饰的Aβ 肽段(如高毒性的pyroglutamate-3 Aβ),提供额外的保护效应[22]。入组轻至中度AD 患者的Ⅰ期临床试验[22]的结果表明,ABvac40 疫苗是安全的,并且可以产生较好的免疫反应;主要的不良反应是头痛、尿路感染和注射部位反应。为进一步评估ABvac40 疫苗的临床效果,入组轻度AD 患者的Ⅱ期临床试验已于2018 年2 月启动,计划于2022 年2 月完成。
2.1.4 ACI-24 疫苗
ACI-24 疫苗(AC Immune 公司研制)由多个Aβ1~15 肽段构成,2 端均连接棕榈酰赖氨酸,旨在模拟Aβ 聚集后的构象。ACI-24 疫苗可以在不激活Aβ 特异性T 细胞应答的情况下引起抗体反应,从而避免中枢神经系统炎症等不良反应[23]。临床前研究的结果显示,ACI-24 疫苗可以通过辅助型T 细胞2 的应答反应,诱导高滴度的抗Aβ IgG2b 抗体[24]。在入组轻至中度AD 患者的Ⅰ期临床试验中,所有试验剂量均是安全的,未发生严重的不良反应,并且观察到高剂量组患者大脑中的淀粉样蛋白减少且认知功能良好。为进一步评估ACI-24 疫苗诱导抗Aβ抗体反应的能力、安全性和耐受性,入组轻度AD 患者的Ⅱ期临床试验已于2018 年6 月启动。
2.1.5 UB-311 疫苗
UB-311 疫苗由2 个合成的N 端Aβ1~14 肽段(B 细胞表位)作为免疫原[25]。将2 个肽段连接到2 个不同外源的辅助性T 细胞表位构成嵌合肽,其目的是增强免疫原性,同时降低佐剂的用量[25]。UB-311 疫苗配制于含有CpG 寡脱氧核苷酸(oligodeoxynucleotide,ODN)的辅助型T 细胞2 导向系统,有利于促进B 细胞免疫反应和调控辅助型T 细胞2 反应,避免辅助性T 细胞1 引起的炎症反应。临床前的体内试验结果表明,UB-311 疫苗未引发小胶质细胞或星形胶质细胞的激活,未发生CD3+、CD4+或CD8+T 细胞浸润[26]。Ⅰ期临床试验的结果显示,UB-311 疫苗的安全性和耐受性良好,在所有的AD 患者中均诱导出高水平的针对Aβ1~10 表位的特异性抗体,其中轻度AD 患者表现出认知功能的改善。由United Neuroscience 公司开展的Ⅱ期临床试验于2018 年8 月启动,旨在评估UB-311 疫苗的免疫原性和患者认知能力的变化,但由于治疗方案不恰当,试验于2019 年10 月提前结束。
2.1.6 其他的第2 代抗Aβ 疫苗
除了前述4 种已获得Ⅰ期临床试验数据的抗Aβ 疫苗以外,Lu AF20513 是由破伤风毒素肽段隔开的3 个Aβ1~12 肽段制成的疫苗,旨在刺激破伤风相关记忆T 细胞产生对Aβ 的抗体反应[27]。这项研究取得了较好的临床前研究成果,其Ⅰ期临床试验已于2019 年5 月结束,但尚未公布结果。
其他的第2 代抗Aβ 疫苗也正在研发之中,如ACC-001、AffitopeAD02 和V950 疫苗,已证实其安全性和耐受性,但并未表现出明显的临床效应,目前相关试验均已停止[28-29]。其中,ACC-001 疫苗以Aβ N 端Aβ1~7 肽段作为免疫原,已经开展了多项入组轻至中度AD 患者的Ⅱ期临床试验,结果表明其安全性和耐受性良好[30-31];然而,治疗组与对照组的认知功能和脑脊液中生物学标志物的水平却无显著差异,这可能与抗体效价不足、免疫原性差、治疗时间短以及样本不足等因素有关[31]。
2.2 Aβ 被动免疫治疗
被动免疫治疗是直接向受体注射靶向Aβ 和(或)其聚合物的单克隆抗体。Aβ 被动免疫治疗的相关研究已有20 多年的历史,最早的研究主要是靶向Aβ 的单体,随后很快出现了靶向Aβ聚合物的抗体。目前,已有多种药物进入Ⅲ期临床试验[32]。尽管有证据表明这些药物可以降低脑脊液中相关生物学标志物的水平,并且减少Aβ斑块,但仍缺乏足够的临床效应。
2.2.1 bapineuzumab 和solanezumab
bapineuzumab 和solanezumab 是最早进入Ⅲ期临床试验[33-34]的药物,2012 年完成的临床试验结果显示这2 种药物均未能改善轻至中度AD 患者的认知功能。bapineuzumab 引起的主要不良反应是淀粉样蛋白相关成像异常(amyloid-associated imaging abnormality,ARIA),其中ARIA-E 是指与血管性水肿和积液相关的MRI 信号改变,ARIA-H 是指与含铁血黄素沉积相关的MRI 信号改变,这可能由Fc 受体介导的小胶质细胞活化所引起[35-36]。ARIA 在APOEε4基因携带者中更为常见,并且呈剂量依赖性[36]。
2.2.2 crenezumab
crenezumab 由Roche 公司于2006 年从AC Immune公司获得许可,是第1 种针对Aβ 寡聚物的抗体[37]。早期的临床试验表明,该药物具有较好的血脑屏障通透性,继发炎症反应较少[38]。尽管crenezumab 未能延缓患者认知能力的下降或改善患者的功能,但由于其显著的安全性和耐受性,crenezumab 进入了临床前期至轻度AD 患者的Ⅲ期临床试验。然而,这项研究已于2019 年终止,因为已获得的研究数据表明患者认知能力的下降并未得到延缓。
2.2.3 aducanumab
aducanumab 是近年来被热议的药物。研究人员通过比较健康老年人与虽有认知障碍但认知衰退进展异常缓慢的老年人的B 细胞库,发现了这种人源性单克隆抗体[39]。aducanumab 是一种IgG1 单克隆抗体,可选择性地与Aβ 聚合物反应,既包括可溶的Aβ 寡聚物,也包括不可溶的Aβ 原纤维[40]。
临床前研究表明,aducanumab 可以透过血脑屏障与靶标相结合,清除转基因小鼠大脑中的Aβ斑块[41]。Ⅰ期临床试验的结果显示,患者大脑中的Aβ 水平显著降低,认知功能衰退减缓,其主要不良反应是ARIA[39]。2015 年,Biogen 公司开展了2 项设计相同的Ⅲ期试验(EMERGE 和ENGAGE),均在出现轻度认知障碍的早期AD 患者中进行,通过PET 扫描证实,这些患者大脑中的Aβ 水平升高;患者分别接受3 种不同剂量aducanumab 的治疗,并且在第78 周试验终点时,采用临床痴呆评分总和量表(Clinical Dementia Rating Scale Sum of Boxes,CDR-SB)评估aducanumab 减缓认知衰退的疗效[42]。2019 年3 月,基于试验中期数据无效性的分析结果,提示aducanumab 达到预期效果的可能性很小,因此Biogen 公司和Eisai 公司宣布终止试验。然而,2020 年10 月Biogen 和Eisai 公司联合宣布将向美国FDA 递交aducanumab 的生物制剂申请。Biogen的申请理由是在EMERGE试验中,接受高剂量aducanumab 治疗的AD 患者的CDRSB 评分与对照组相比降低了23%,获得了统计学意义上的显著改善;但是,接受低剂量治疗的AD患者的CDR-SB 评分变化没有达到统计学意义[43]。然而,在试验设计完全相同的ENGAGE 试验中,无论是高剂量还是低剂量aducanuma,均未显示出对患者的认知能力有显著改善[43]。美国FDA 于2021年6 月7 日通过加速审批通道批准了aducanumab治疗AD 的申请,理由是PET 证明aducanumab 可以减少脑内的Aβ 沉积。随后,在2021 年7 月8日,aducanuma的适应证范围被限于轻度AD。然而,由于aducanumab 在前期临床试验中获得了矛盾的结果,因此对于该药物的安全性和有效性仍存有疑虑[44-46]。根据加速审批规定,Biogen 公司需要在aducanumab 上市后继续开展Ⅳ期随机对照试验以验证其临床效果,并且要求在2030 年2 月提交最终报告;如果不成功,其获批申请可能会被撤销[47]。
2.2.4 gantenerumab
gantenerumab 是由Roche 公司开发的IgG1 单克隆抗体,是第1 种进入AD 临床研究的完全人源性单克隆抗体[48]。gantenerumab 可与大脑中聚集的Aβ 相结合,通过募集小胶质细胞和激活的巨噬细胞,降解沉积的Aβ[48]。2010 年,Roche 公司启动临床前期AD 患者的Ⅱ期临床试验以检验gantenerumab 的临床疗效,并且在2012 年将这项临床试验扩展为Ⅱ/Ⅲ期临床试验,即Scarlet RoAD 试验[23]。研究的第1 部分,gantenerumab的治疗剂量为每4 周105 或225 mg,共治疗104周,试验终点是CDR-SB 评分和大脑Aβ 水平的变化;研究的第2 部分,在104 周疗程结束后继续维持原方案治疗2 年。至2014 年,有50%的患者完成了研究的第2 部分,但是基于试验中期数据的无效性分析结果,预测达到预期效果的可能性很低,Roche 公司终止了Scarlet RoAD 试验[49]。然而,至2015 年,生物学标志物和疗效分析的结果显示,在疾病快速进展的AD 患者中存在gantenerumab的剂量依赖效应,因此认为可能需要使用更高剂量的ganteneurumab 才能达到临床获益。基于这一发现,Scarlet RoAD 试验于2017 年转为开放性试验,在研究的第3 部分改用高剂量ganteneurumab(每4 周1 200 mg)连续治疗3 年,并于2020 年9 月结束研究,结果显示高剂量ganteneurumab可在2 年内有效去除Aβ 斑块[50]。目前正在进行的临床试验包括ganteneurumab 对早期AD 患者的疗效和安全性的研究以及AD 患者长期接受ganteneurumab 治疗的安全性和耐受性研究。这2项研究均将于2023 年结束。
2.2.5 lecanemab(BAN2401)
lecanemab 是Biogen 公司另一种进入Ⅲ期临床试验的Aβ 被动免疫治疗药物。lecanemab 是小鼠单克隆抗体mAb158 人源化生成的IgG1 单克隆抗体,维持了mAb158 的特性,能够高度选择性地与Aβ 原纤维结合[51]。
Ⅰ期临床试验证实,这种人源化IgG1 抗体没有增加ARIA-E/H 的风险[52]。Ⅱb 期临床试验采用贝叶斯设计,对856 例轻度认知障碍AD 患者进行了18 个月的研究,结果显示患者大脑中Aβ 的减少和认知功能的改善有统计学意义,并且呈剂量依赖性,仅<10%的患者出现ARIA[52]。Ⅱb 期扩展临床试验以最高剂量(每2 周10 mg/kg)重新入组患者,并且持续2 年,目的是进一步评价药物的安全性。一项名为“Clarity AD”的Ⅲ期临床试验于2019 年3 月开展,通过评估早期AD 患者接受lecanemab 治疗(每2 周10 mg/kg)18 个月后CDR-SB 评分的变化以评估药物的疗效、安全性和耐受性;扩展阶段将观察治疗作用是否能够长久维持。
除上述进入Ⅲ期临床试验的药物,还有其他一些抗Aβ 抗体也进入了临床试验阶段(表1),如GSK933776、donanemab(LY3002813)和ponezumab[53-56]。这些抗Aβ 抗体可以靶向多种形式的Aβ,包括Aβ 单体、寡聚物和原纤维,其作用机制涉及激活小胶质细胞以促进细胞外Aβ的清除,以及阻断初级和次级Aβ 的成核作用以阻断Aβ 在大脑中的沉积[57]。
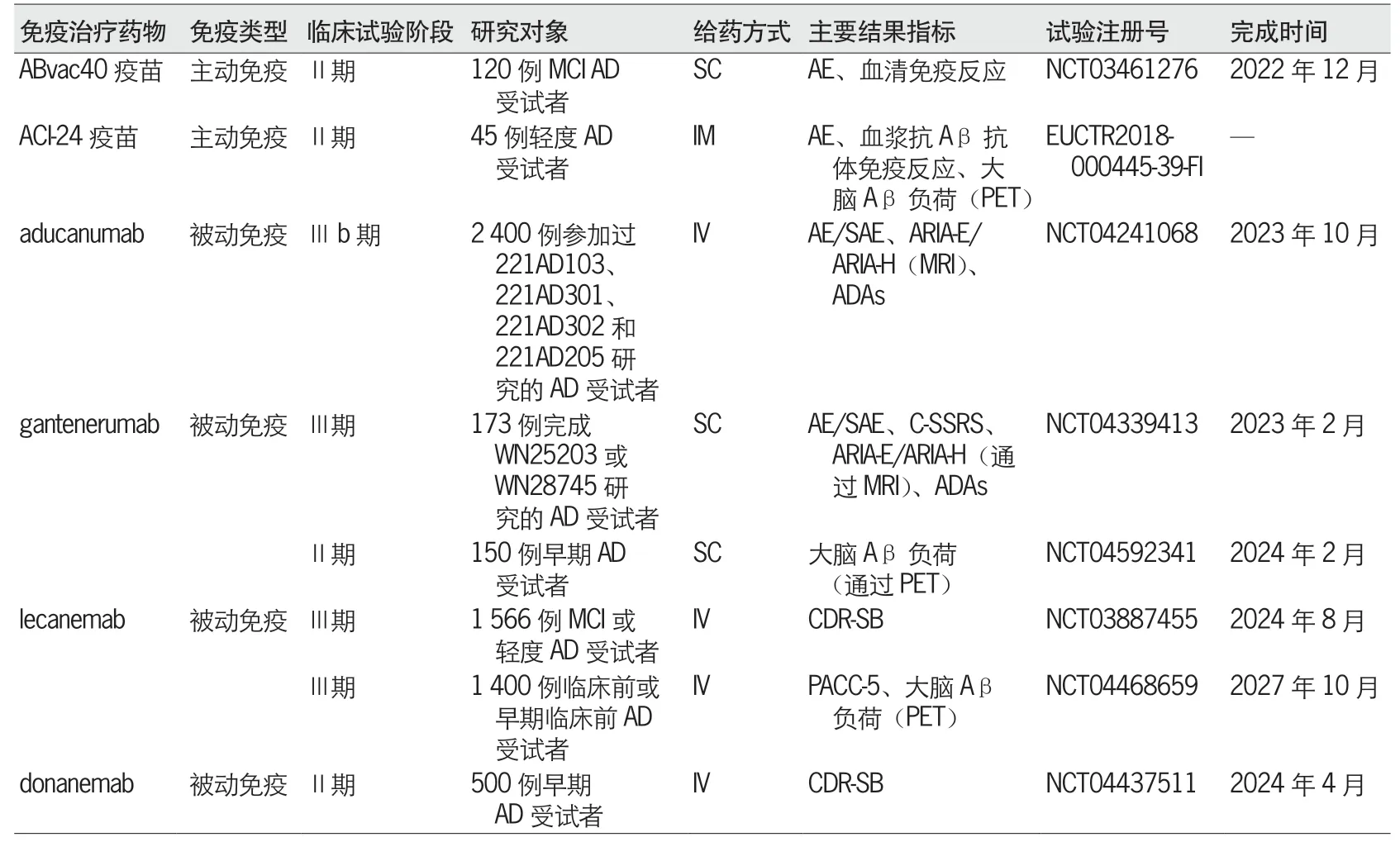
表1 正在进行临床试验的靶向Aβ 的免疫治疗药物
3 结语
总之,当前AD 临床试验中的靶向Aβ 免疫治疗的药物尚未达到缓解认知功能减退的预期目标。寻找靶向Aβ 的新疗法的主要理论依据是淀粉样蛋白级联假说,即假定Tau 蛋白引起的NFTs 是Aβ产生和清除失衡的下游产物。最初认为,AD 的神经变性是由淀粉样斑块中的Aβ 引起的,后来才发现主要是可溶性Aβ 寡聚物的作用。多年来,大多数进入临床试验的药物系靶向Aβ,但都难以获得理想的结果,许多进入Ⅲ期临床试验的项目宣告失败;但在2021 年,一种抗Aβ 药物(aducanumab)成功获得美国FDA 的批准,成为首个针对AD 病理学基础进行免疫治疗的药物。虽然近年来新开发的靶向Aβ 药物较以往有所减少,但其在AD 后期临床试验中所占的比例仍然很高(约为40%)。
近年来,靶向Tau 蛋白的免疫治疗药物也逐渐进入Ⅰ期和Ⅱ期临床试验,并且已有研究发现,Tau 蛋白与AD 患者临床认知障碍的相关性更好[58]。2020 年11 月,第13 届阿尔茨海默病临床试验(Clinical Trials on Alzheimer’s Disease,CTAD)会议上有报告指出,TAURIEL Ⅱ期试验中semorinemab(一种靶向Tau 蛋白的抗体)没有改善临床前期或早期AD 患者的预后,似乎预示着靶向Tau 蛋白的免疫治疗之路也将充满坎坷。另外3 种Tau 蛋白抗体(ABBV-8E12、gosuranemab 和zagotenemab)将 于2021 年获得疗效方面的数据。Aβ 免疫治疗的起步较早,进展更为深入;而Tau 蛋白免疫治疗的起步较晚,目前的临床试验大多处于验证药物安全性和耐受性的阶段。因此,在推进Tau 蛋白免疫治疗之前,有必要先总结Aβ 研究的经验和教训。
AD 的治疗关键是早期治疗甚至是预防性治疗。Aβ 免疫治疗的结果显示,一旦出现认知障碍等症状,大脑中神经元的病理变化是不可逆的,因此导致临床疗效有限。针对Aβ 治疗的临床试验也大多选择临床前期、早期和中期的AD 患者。评价药物的疗效不应只关注临床症状是否得到改善,还应关注症状有否加重。研究表明,AD 的分子病理学改变早于任何临床症状的出现,并且在体内产生类似病毒样的传播。因此,通过检测相关生物学标志物,实现临床症状前的AD 早期诊断,并进行精准分期,显得尤为重要。
就被动免疫治疗而言,多项失败的临床试验(如aducanumab、gantenerumab)让人们关注到药物剂量不足的问题。正常情况下,血清中<0.1%的免疫球蛋白能够穿过血脑屏障,因此确定药物的有效剂量显得尤为重要[57]。一些临床前试验也针对促进药物的脑吸收,例如利用转铁蛋白受体介导的胞吞作用[59]。
联合疗法被认为是免疫治疗未来的发展方向。已证实AD 的病理过程涉及多种生化途径的改变,包括Aβ 和Tau 蛋白,尤其是Aβ 聚集形成的寡聚物和原纤维存在多态性,导致单个抗体的作用有限,而联合治疗能够实现单药治疗无法达到的效果,但随之而来的问题是如何确定联合治疗中各种药物的组成和剂量,均有待进一步验证。
随着aducanumab 获得美国FDA 的批准,免疫治疗AD 的基础和临床研究均进入了新阶段。随着人们对AD 发病机制的深入研究,将有更多靶向AD 的药物进入临床试验。近年来,已发现更多的生物学标志物,可以实现AD 的早期诊断;此外,开发针对Aβ 和Tau 蛋白等靶点的联合疗法,将为AD 治疗提供更多新的思路和手段。
