基于微生物组大数据搜索的疾病检测
2021-09-06张玉凤荆功超李劲华苏晓泉
张玉凤 荆功超 李劲华 苏晓泉
微生物是地球上最早进化形成的生命体之一,它们与后续进化的动植物以及人类等相互影响,形成各种各样的复合体系。微生物在自然生态圈中几乎无处不在,它们绝大多数并不是孤立的,而是以“微生物群落” (亦称菌群)的形式共存。通常,也用“微生物组”(microbiome)来表示某个特定环境或者生态系统中全部的微生物及其遗传信息。
人体微生物组被称为人的“第二基因组”。 作为与生俱来又无处不在的“小伙伴”,微生物组与我们有着千丝万缕的关联[1]。随着生物技术与大数据的发展,研究发现人体菌群与人的健康状况的变化和疾病的发生发展密切相关,如炎症性肠病[2]、Ⅱ型糖尿病[3]、结直肠癌[4]等疾病的发生都伴随着肠道菌群的改变。微生物组检测具有非侵入性、可量化性、可预警等优势,例如,儿童的口腔菌群检测仅需几滴唾液,就可以提前预测龋齿的患病风险[5],使其在疾病识别、治疗方案制定、预后评估等方面有着巨大的潜力。因此,基于微生物组的健康状态检测一直是精准医学和大健康领域的热点问题之一。那么,如何用菌群判断和区分疾病呢?
微生物组疾病检测的方法与不足
一个微生物群落中可能存在成百上千种不同的物种。因此,微生物组检测首先用DNA测序和生物信息分析来获取菌群的结构成分,也就是知道其中有什么微生物,含量比例是多少。之后,常规的做法是将某类疾病患者的菌群样本和健康样本进行对比,发现其中关键的差异物种作为生物标记(bio-marker),并根据标记的含量高低来判断待检样本是否罹患该类疾病。然而,这类方法在适用广泛性、疾病特异性、污染抵抗性等方面有着明显的不足,从而阻碍了微生物组技术的进一步应用。
首先,生物标记法在疾病普适程度上限制大,易出现“漏诊”问题。利用某类疾病样本和健康样本中具有差异的物种作为生物标记,检测范围也就仅限于该疾病类型,否则将无法正确地识别出样本的状态。然而在实际的检测情况中,不同人群中同一疾病的生物标记物种往往不一致[6],甚至许多疾病沒有明确的标记。当样本的疾病超出模型的检测范围时,很难综合地判断待测样品是否健康。
同时,生物标记法在检测特异性上的缺陷会导致“误诊”问题。前期研究表明,已知的每一种生物标记均会与至少两种不同的疾病存在关联[7]。因此,生物标记的检测范围较大,涵盖的疾病类型较多(例如5个以上)时,在区分不同疾病的过程中会容易出现误判。
此外,生物标记容易受到外来“污染物”的干扰,从而影响检测结果。由于我们身体上和生活的环境中几乎所有可及之处都有微生物的存在,检测的过程中可能会混入来自身体其他部位或者环境中的微生物,这些“污染物”会改变菌群成分分析中生物标记的相对含量,从而降低疾病识别的稳定性。
微生物组搜索引擎和大数据挖掘为疾病检测提供新思路
随着DNA测序成本的降低和微生物组研究的迅猛发展,菌群的测序数据正在以前所未有的速度累积。然而,每当新的微生物组数据产生时,受限于该领域大数据分析工具的匮乏,难以与原有的海量数据进行快速的比对,束缚了对新数据的认识与解读。笔者团队成功开发了微生物组搜索引擎(Microbiome Search Engine,以下简称搜索引擎),通过物种成分分类的动态索引机制和Meta-Storms比对算法[8],实现了微生物群落整体层面的高速搜索,从而在急速增长的已知数据中,迅速准确地找到与新样本在群落结构上高度相似的匹配样本,使得未知菌群的“温故而知新”成为可能。
对于每一个新的微生物群落,利用搜索引擎在已有的数据库中进行搜索,根据最佳的匹配度来计算的“微生物组新颖指数”(以下简称新颖指数),能够量化出该样本的新奇性和独特性。新颖指数值越高说明样本与先前已大规模采样收录的菌群相似性越低,也就越独特。研究人员通过对2010—2017年产生的来自全球不同地域、不同环境的共100 000多例微生物组样本进行追踪分析,结果显示人体菌群的新颖指数变化在全局范围内是收敛性发展,且已经呈现接近饱和的状态[9],而自然环境菌群的收敛性却不明显。人体微生物组的这种收敛性也称为 “搜索边界效应”,即虽然人与人之间菌群结构和组成差异很大,但从整体尺度来看,其变化总在一定的范围之内。
当进一步缩小范围,健康的人体微生物组是不是也存在类似的“搜索边界效应”,使得健康菌群的新颖指数总在一定正常范围之内,而疾病菌群明显高于正常水平,从而利用搜索的策略来实现对非正常状态菌群的筛选和识别。研究人员再次利用多个地区超过15 000例正常人群的微生物组作为基准,发现多种不同疾病样本的新颖指数均要显著高于健康样本,从而验证了健康微生物组的“搜索边界效应”,并为疾病检测提供了前提。
基于大数据搜索的微生物组疾病检测
与现有的生物标记法相比,基于搜索的疾病检测不再依赖疾病相关的特定标识微生物,而是利用待测菌群的整体组成结构与已有数据的相似度来实现检测[10]。此方法分为两步:第一步,计算待测样本相对于健康数据库的新颖指数,根据其异常程度即可评估其是否健康;第二步,与疾病数据库进行比对,根据最佳匹配结果即可识别具体的患病种类,从而依次回答“是否健康”和“哪种疾病”这两个问题。
经过3000余例人体肠道微生物组样本的检测表明,针对炎症性肠病(inflammatory bowel disease, IBD)、结直肠癌(colorectal cancer, CRC)、艾滋病毒感染(human immunodeficiency virus,HIV)和肠腹泻病(enteric diarrheal disease,EDD)这4种疾病,搜索引擎在回答“是否健康”和“哪种疾病”这两个问题上的准确率均超过75%,显著高于目前常用的生物标记法,从而有效地降低了“漏诊”和“误诊”的可能。此外,由于炎症性性病和肠腹泻病具有大量相同的生物标记,导致生物标记法容易产生混淆,降低了对二者的区分度和识别率,从而产生了“短板效应”,而搜索引擎对其中每一种疾病的检测准确性都较为一致。
基于搜索的检测方法对微生物污染具有较强抵抗性
一般来讲,科学实验数据的采样、封存、测序等过程由专业的操作人员按照标准工作流程来完成,会在极大程度上降低污染的可能性。而当未来菌群检测逐渐普及后,菌群的采样会由用户或患者自行来完成。例如,当我们通过棉签涂抹手部来获得皮肤菌群时,可能会难以避免地混入其他身体部位或者生活环境当中的污染微生物,从而干扰检测结果。为了测试不同检测方法对污染物的抵抗性,研究人员在测试数据中分别混入5%~20%不同比例的室内环境微生物作为“污染”。结果显示,即便在最高的污染率下,搜索引擎仍具有71%以上的良好准确度。但由于高污染会显著改变群落中物种的相对比例,生物标记法检测结果受影响较大,平均识别率已然骤降到了60%左右。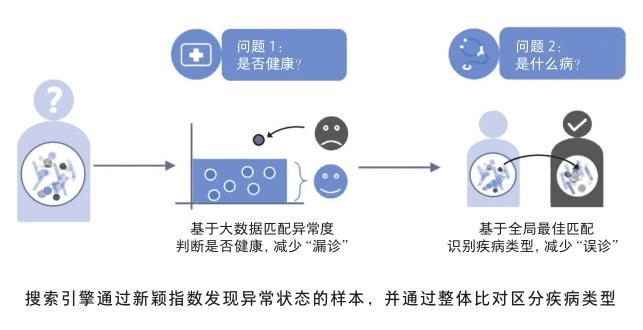
以多角度、大数据分析为特征的微生物組研究时代已经来临[11,12]。得益于交叉学科研究,配合微生物组高通量测序和高性能计算方法学等方面迅猛的进步,以大数据驱动为主的微生物组创新研究模式将会为微生物组技术在人体健康领域的研究和临床应用带来突破性新手段和新思路,深化对共生菌群和人类健康之间互作关系的认识,造福社会,造福人类,造福自然。
[1]Turnbaugh P J, Ley R E, Hamady M, et al. The human microbiome project: exploring the microbial part of ourselves in a changing world. Nature, 2007, 449(7164): 804-810.
[2]Halfvarson J, Brislawn C J, Lamendella R, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nature Microbiology, 2017, 2(5): 17004.
[3]Qin J J, Li Y R, Cai Z M, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature, 2012, 490(7418): 55-60.
[4]Wirbel J, Pyl P T, Kartal E, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specifi c for colorectal cancer. Nature Medicine, 2019, 25(4): 679-689.
[5]Teng F, Yang F, Huang S, et al. Prediction of early childhood caries via spatial-temporal variations of oral microbiota. Cell Host & Microbe, 2015, 18(3): 296-306.
[6]Duvallet C, Gibbons S M, Gurry T, et al. Meta-analysis of gut microbiome studies identifi es disease-specifi c and shared responses. Nature Communications, 2017, 8(1): 1784.
[7]Jackson M A, Verdi S, Maxan M E, et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nature Communications, 2018, 9(1): 2655.
[8]Su X, Xu J, Ning K. Meta-Storms: Efficient search for similar microbial communities based on a novel indexing scheme and similarity score for metagenomic data. Bioinformatics, 2012, 28(19): 2493-2501
[9]Su X, Jing G, McDonald D, et al. Identifying and predicting novelty in microbiome studies. mBio, 2018, 9(6): e02099-18.
[10]Su X, Jing G, Sun Z, et al. Multiple-disease detection and classification across cohorts via microbiome search. mSystems, 2020, 5(2): e00150-20.
[11]Su X, Jing G, Zhang Y, et al. Method development for crossstudy microbiome data mining: challenges and opportunities. Computational and Structural Biotechnology Journal, 2020, 18: 2075-2080.
[12]Taroncher-Oldenburg G, Jones S, Blaser M, et al. Translating microbiome futures. Nature Biotechnology, 2018, 36(11): 1037-1042.
关键词:微生物组 大数据 搜索引擎 疾病检测 生物信息 ■
