1,3,4-噁二唑联呋咱含能化合物的设计及构效关系理论研究
2021-09-06毕福强王子俊张家荣吴敏杰王伯周
薛 琪,毕福强,王子俊,廉 鹏,张家荣,吴敏杰,王伯周
(1.西安近代化学研究所,陕西 西安 710065;2.氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065)
引 言
随着武器弹药使用环境的日益苛刻,全球弹药安全事故频出[1],对于新型高能不敏感材料(爆速大于9000m/s,撞击感度大于15J)的需求愈发紧迫[2-3]。然而,对于含能化合物分子,能量与感度存在固有矛盾:通常含能化合物分子需要引入较多的致爆基团(如硝基)提高生成焓和氧平衡值,而这种强吸电子基会导致分子内部电荷分离,电荷分离程度越高,分子中C—NO2/N—NO2键越弱,分子越不稳定[4-6]。
研究人员认为,氮杂芳环构建稳定的共轭分子骨架可有效降低致爆基团导致的分子电荷分离程度[6],是获得高能不敏感含能化合物的有效途径之一,其中,五元氮杂芳环噁二唑环(呋咱、1,2,4-噁二唑和1,3,4-噁二唑)因具有高稳定性、高氮氧平衡值受到广泛关注。理论计算结果表明,利用不同的噁二唑环异构体可共同构建稳定性良好的分子骨架[7]。事实证明,迄今为止合成的1,3,4-噁二唑联呋咱类含能化合物普遍具有热稳定性和安全性能优良的特点[8-10],如4,4′-二(5-氨基-1,3,4-噁二唑-2-基)-3,3′-偶氮呋咱(化合物2),分解温度309℃,撞击感度40J,爆速8458m/s,在类似氮杂环结构化合物中,如二氨基偶氮呋咱(DAAzF)、4,4′-二四唑-3,3′-偶氮呋咱(AzTF),综合性能突出,表明1,3,4-噁二唑联呋咱是优良的含能化合物共轭分子骨架。然而,由于1,3,4-噁二唑为缺电子环,其2,5-位氨基很难氧化成高能基团硝基[8-10],爆轰性能与高能不敏感含能化合物的要求尚存差距。为了弥补能量不足的缺陷,理论上可以在1,3,4-噁二唑3,5-位的α-碳上引入高能基团(如三硝基甲基和氟代偕二硝基),进一步提升化合物的爆轰性能。然而,此类化合物的设计、爆轰及安全性能的研究均未见报道,1,3,4-噁二唑联呋咱分子骨架与高能量密度基团爆轰性能与安全性能间的构效关系尚不清晰。
基于此,本研究基于1,3,4-噁二唑联呋咱分子骨架,设计了一系列1,3,4-噁二唑联呋咱高能化合物,采用量子化学密度泛函理论研究了母体结构和含能基团对化合物结构与性能的影响,期望获得性能优异的含能化合物分子结构,同时,也为后续开展相关研究工作提供理论依据。
1 计算方法
1.1 分子结构式
为探讨1,3,4-噁二唑联呋咱分子骨架与高能量密度基团爆轰性能与安全性能间的构效关系,本研究设计的1,3,4-噁二唑联呋咱化合物的分子结构如图1所示。根据致爆基团及分子骨架中杂环个数和联接方式不同,将其分为A、B、C、D和E5类。
1.2 爆轰及理化性能的计算方法
运用Gaussian 09软件[11]中B3LPY/6-31G(d, p)基组[12]对各分子进行优化。采用原子化方案,使用CBS-4M模型[13]以获得较精确的气相生成焓值。绝大多数含能材料在常温常压下为凝聚态,因此要准确计算爆轰性能需要知道其固相生成焓。固相生成焓根据Hess定律计算得到,其计算公式为:
(1)
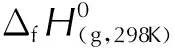
(2)
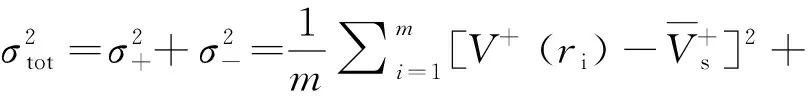
(3)
(4)
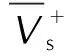
在优化构型基础上,用Monte-Carlo法[15]计算法在Multiwfn程序下计算分子的理论摩尔体积(Vm),为了减小误差,取100次计算值的平均值。共价型含能化合物的理论密度为分子量和理论摩尔体积的比值:
(5)
在得到密度生成焓等相关数据后,采用的EXPLO5(V6.04)爆轰性能计算软件[16]计算出理论爆速和爆压等参数。
对于系列结构和热解机理相类似的爆炸物,断裂某键所需能量越少,表明该键强度越弱,越易成为热解引发键,相应的化合物越不稳定,预测其感度可能越大[17]。分子中A—B的键离解能(BDE)指均裂该键所需的能量,通常用经零点能矫正的产物与反应物的能量之差表示。对下列均裂A—B键的反应为:
RA—BR′(g)→RA·(g)+R′B(g)
(6)
其键离解能为:
BDE(RA—BR′)=[E(RA·)+E(R′B·)]-E(RA—BR′)
(7)
式(7)中:RA—BR′为反应物;RA和R′B为均裂后所得产物(自由基)。
2 结果与讨论
2.1 稳定构型分析
10种1,3,4-噁二唑联呋咱含能化合物的稳定构型如图2所示。键长如表1所示。
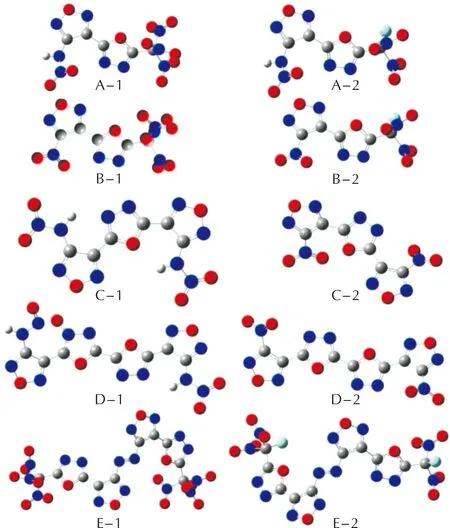
图2 A~E系列化合物的稳定几何构型Fig.2 Geometiric configuration of A—E compounds
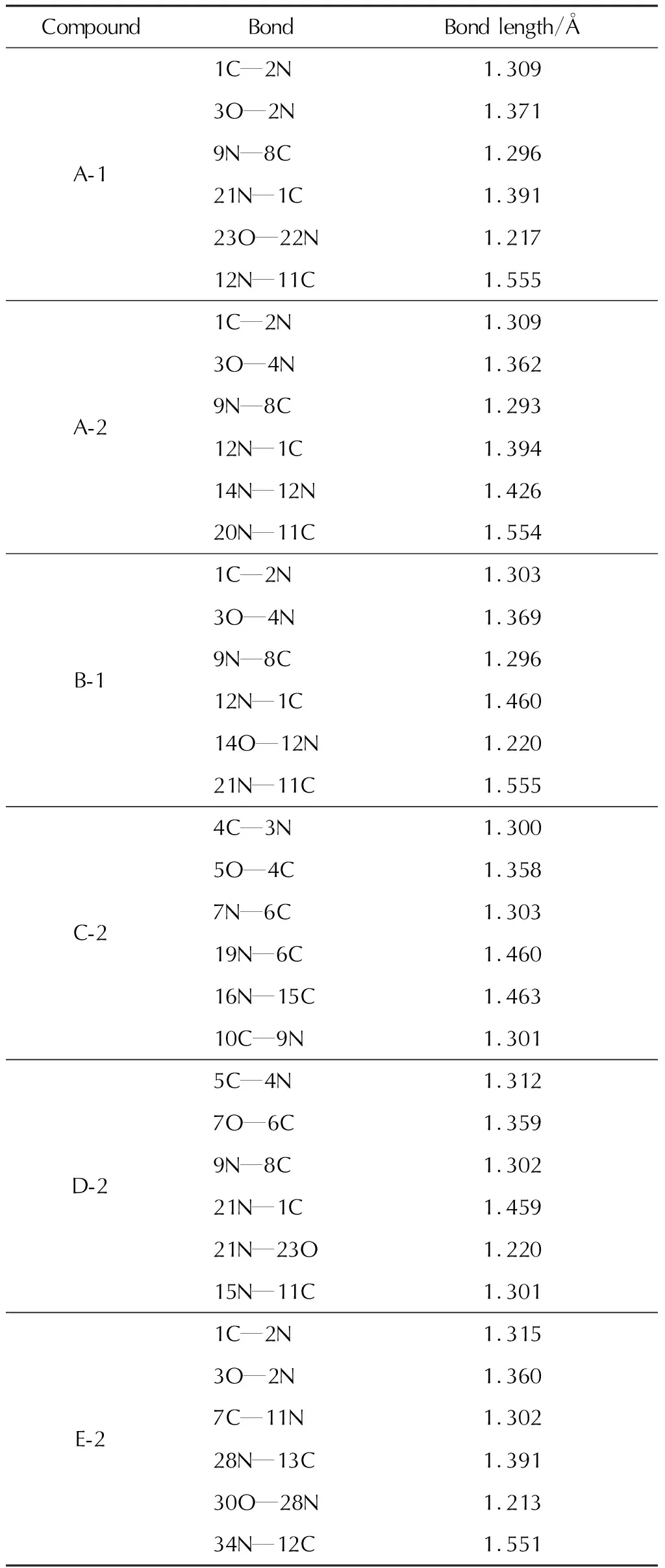
表1 部分模型化合物的键长Table 1 Bond lengths of model compounds
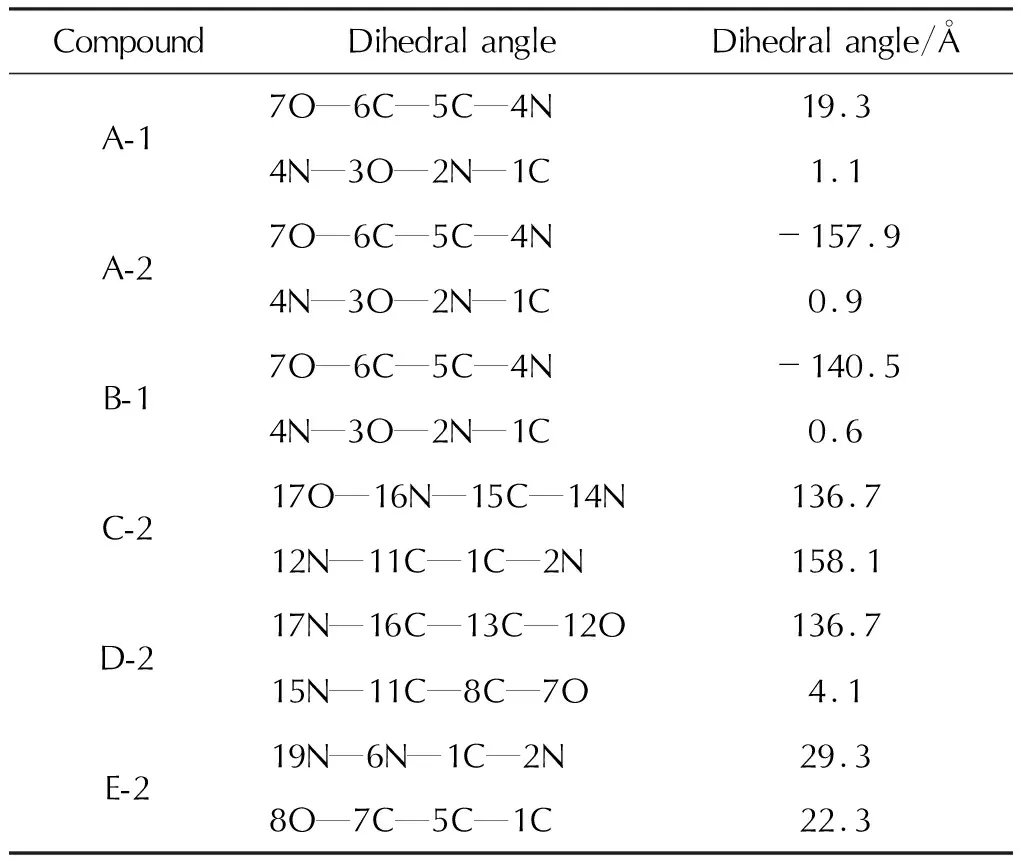
表2 部分模型化合物的二面角Table 2 Dihedral angles of model compounds
2.2 能量性能
对10种1,3,4-噁二唑联呋咱化合物的理论密度(ρ)、固相生成焓(ΔHf(s))、氧平衡(OB)、理论爆速(D)、理论爆压(p)进行了计算,结果见表3。

表3 1,3,4-噁二唑联呋咱含能化合物的能量性能Table 3 Energetic performances of furazan-1,3,4-oxadiazole-based energetic compounds
化合物的固相生成焓越高,其分解后释放的能量越高,有助于提高化合物的爆热。由于呋咱环和1,3,4-噁二唑可提供给分子较高的生成焓,因此,在这10种含能化合物中,以多噁二唑环为骨架组成的分子结构固相生成焓较高,总体而言E>D>C>A/B,其中,化合物E-1的生成焓最高,为662.02kJ/mol。在母体骨架相同时,含能基团与生成焓的大小关系为:三硝基甲基>氟代偕二硝基,硝基>硝氨基。
密度、爆压和爆速是衡量含能化合物的能量的最直接的3项参数。本系列化合物的理论密度介于1.89~1.991g/cm3之间,多种化合物密度超过1.90g/cm3,总体而言B>E>A>C≈D。结果表明,氟代偕二硝基及三硝基甲基的引入可显著提高化合物密度,其中,氟代偕二硝基对化合物密度提升最大,三硝基甲基次之。化合物爆速和爆压受其生成焓、密度、分子组成影响,因此,引入三硝基甲基和氟代偕二硝基含能基团的化合物爆速更高。
10种化合物氧平衡值介于-36.5%~-2.3%之间。各系列纵向对比可以看出,A系列氧平衡最高,氧平衡值与硝基含量成正相关。本系列共有5种化合物理论爆轰性能优于HMX,分别为A-1、B-1、B-2和E-1,均为引入三硝基甲基的化合物,可见,在1,3,4-噁二唑联呋咱骨架上构建三硝基甲基,是一种设计合成高能量含能化合物的有效途径。
2.3 静电势分析
分子的静电势与分子间的相互作用息息相关。据文献报道[19-21],在相同的计算方法和电子密度等值面上,通过Multiwfn 3.0.1程序[22-23]可对正负静电区域的面积和强度进行统计,结合可视的静电势图对化合物的感度性能进行比较和定性预测。
在B3LPY/6-31G(d, p)水平下,得到了化合物在0.001 electron bohr-3电子密度等值面上的三维静电势示意图,见图3,并对正负静电势区域的面积和强度进行了统计,见表4。

表4 1,3,4-噁二唑联呋咱含能化合物的静电势参数Table 4 Electrostatic potential parameters of furazan-1,3,4-oxadiazole-based energetic compounds
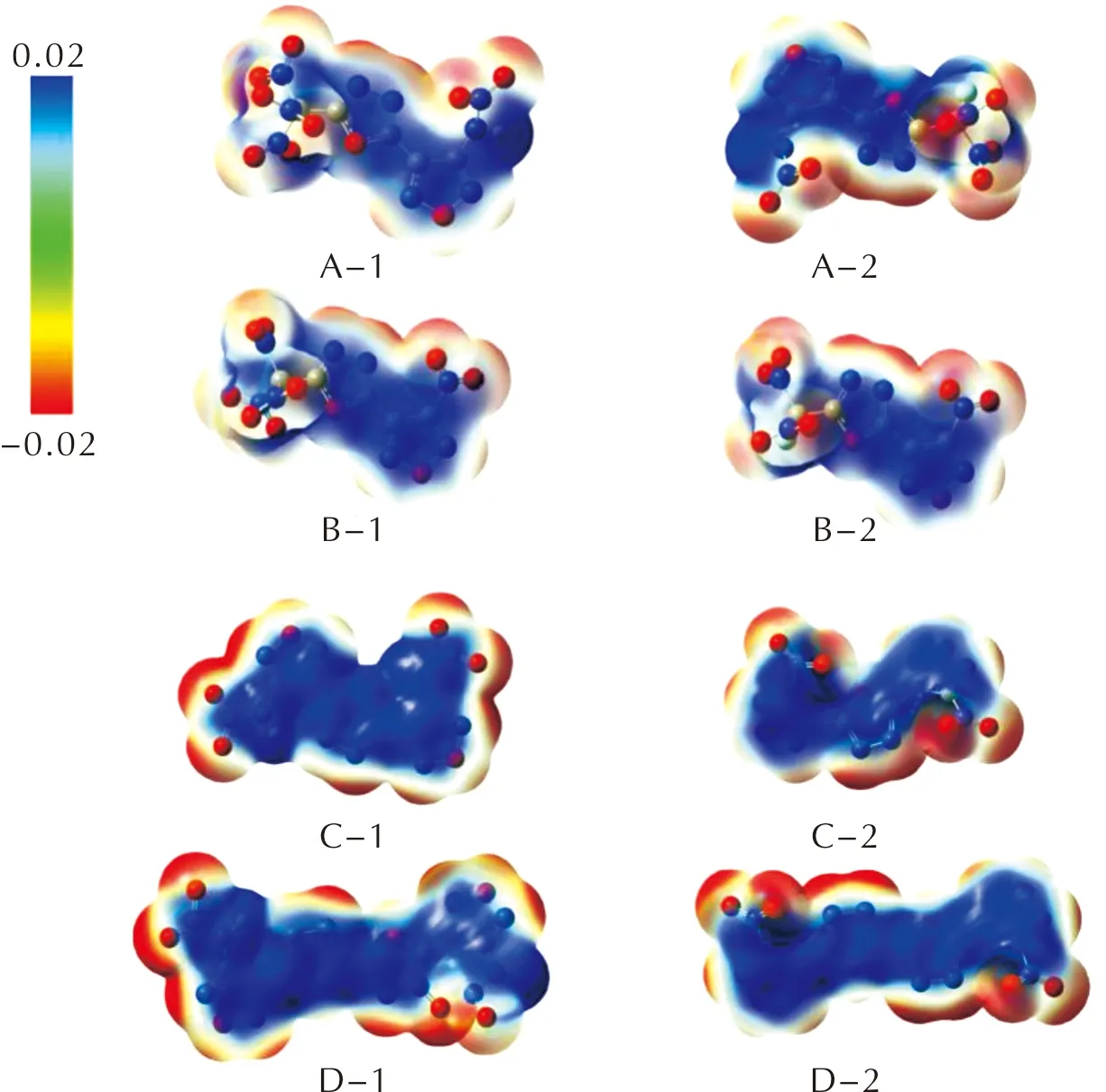

图3 1,3,4-噁二唑联呋咱化合物的静电势图Fig.3 Electrostatic potential distributions of furazan-1,3,4-oxadiazole-based energetic compounds
从图3中可以看出,化合物的负静电势主要分布在化合物骨架的呋咱环N—O—N、1,3,4-噁二唑的N—N及含能基团的硝基氧上,正静电势主要分布在呋咱、1,3,4-噁二唑的中心环以及氨基氢、碳原子、碳上氢原子上。Klapotke等[19,21]认为,在含能体系中,正电荷区域面积大于负电荷,当正负电荷面积比(As+/As-)接近1.5时化合物感度较高。本系列所有分子的As+/As-值位于1.0 ~ 1.4区间,与含能化合物静电势分布特征相符。
2.4 键离解能
化学键的键离解能(BDE)越低,其在化合物在分解过程中首先发生断裂的可能性越高。本系列10种化合物的分子结构和热解机理比较相似,因此可用BDE判别其稳定性和感度的相对大小。为了系统探究含能基团与本系列1,3,4-噁二唑联呋咱化合物稳定性的构效关系,本节计算了分子内所包含的三硝基甲基内的C—NO2(a)、氟代偕二硝基内的C—NO2(b)、1,2,5-噁二唑与硝基间的C—NO2(c)键、1,3,4-噁二唑与三硝基甲基间的C—C(a)键,1,3,4-噁二唑与氟代偕二硝基间的C—C(b)、1,2,5-噁二唑与硝氨基间的C—N(d)以及氟代偕二硝基内的C—F、硝氨基内的N—NO2的键离解能(BDE),经自由键级轨道(NBO)分析,最小键级包含在它们之内,结果列于表5中。
由表5可见,该系列化合物的BDE均大于100kJ/mol,满足高能材料稳定性要求(BDE=80~120kJ/mol)。含能基团的BDE大小平均值的顺序关系是C—N(a) 表5 含能基团全部化学键的键离解能(BDE)和最弱键键长Table 5 The weakest bond length and BDE of energetic groups 与母体骨架为2,5-二呋咱-1,3,4-噁二唑的C系列化合物相比,在取代基相同的情况下,共轭骨架中多引入一个1,3,4-噁二唑环的D系列化合物,BDE均大幅提高,其中D-2最弱键的BDE为263.97kJ/mol,略低于TATB(287.2kJ/mol),大幅优于RDX(145.6kJ/mol),结合其良好的爆轰性能(密度1.908g/cm3,爆速8742m/s),有望成为不敏感含能材料。 由此可见,在含有1,3,4-噁二唑的大共轭母体骨架上引入高能量基团是设计不敏感含能化合物的有效途径。 (1)三硝基甲基和氟代偕二硝基可有效提高含能化合物的爆轰性能,但C—NO2较易成为分解过程中的引发键,将其引入1,3,4-噁二唑联呋咱的大共轭母体骨架上,其最弱键键离解能明显升高,可有效提高其稳定性,是设计高能不敏感含能化合物的有效途径。 (2)理论计算结果表明,D-2最弱键的BDE为263.97kJ/mol、密度1.908g/cm3、爆速8642m/s,是潜在的不敏感含能材料;E-1密度为1.969g/cm3、爆速达9130m/s、爆压38.82GPa,爆轰性能与HMX相当,且最弱键BDE为131.6kJ/mol,有望成为一种性能优良的高能量密度化合物。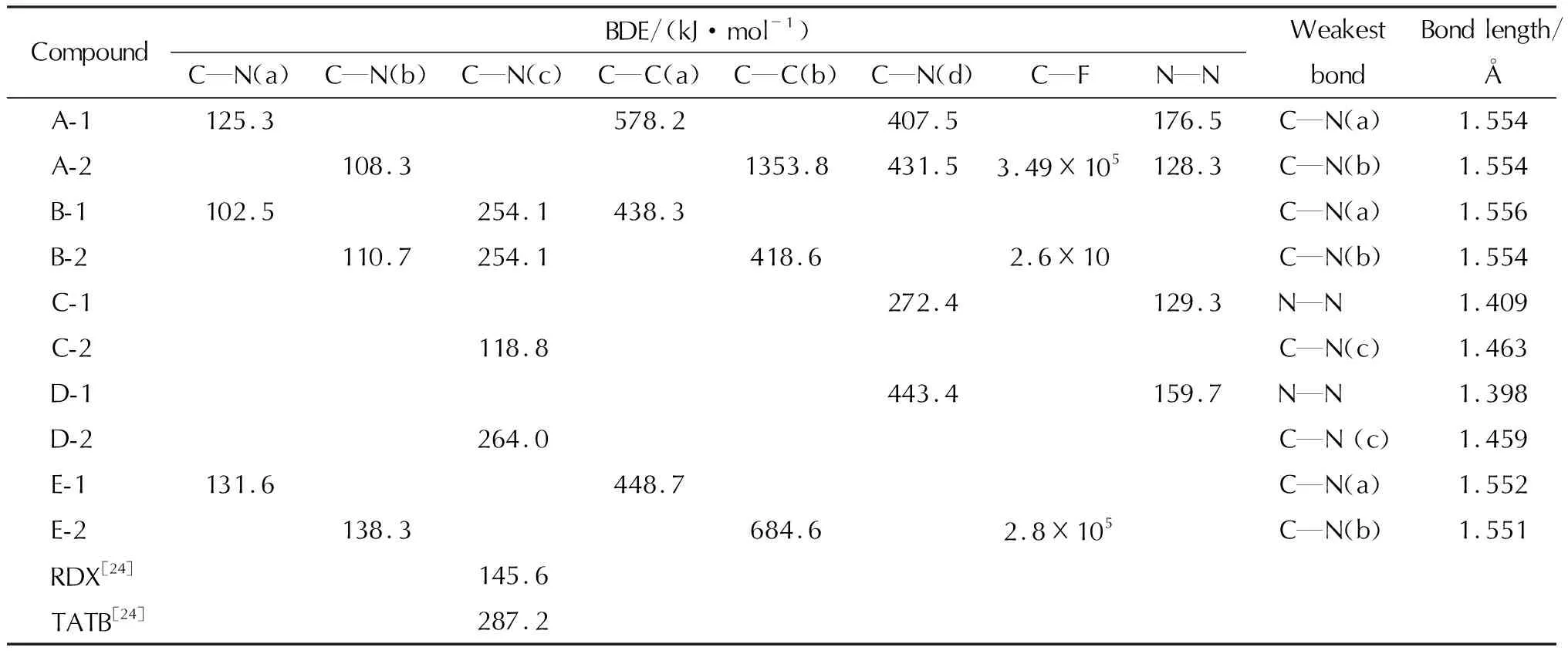
3 结 论
