新型非异氰酸酯固化体系研究进展
2021-09-06卢先明王晓川莫洪昌徐明辉
刘 宁,卢先明,王晓川,莫洪昌,张 倩,李 辉,徐明辉
(1.西安近代化学研究所,陕西 西安 710065;2.氟氮化工资源高效开发与利用国家重点实验室,陕西 西安 710065;3.华南理工大学 生物医学科学与工程学院,广东 广州 510006)
引 言
由黏合剂、固化剂以及各种网络调节剂组成的聚合物网络构成了复合固体推进剂的基本“骨架”,对其加工性能、力学性能和安全性能有着重要影响[1-2]。因此,由黏合剂与固化剂组成的固化体系是构成复合固体推进剂的关键组分之一,更是其更新换代的标志。
在固体推进剂现役固化体系中,应用最广泛的是羟基/异氰酸酯固化体系,其典型代表为端羟基聚丁二烯(HTPB)/甲苯二异氰酸酯(TDI)[3]。然而异氰酸酯固化体系存在以下不足[4-5]:(1)异氰酸酯除了与羟基反应形成氨酯键外,还容易与空气或固体组分携带的水反应,生成二氧化碳气体,导致药柱产生气孔和裂纹;(2)异氰酸酯毒性较大,对环境、人体危害性大;(3)异氰酸酯固化体系固化温度较高(65~75℃),药柱冷却到室温后产生收缩应力,易产生脱粘现象;(4)异氰酸酯固化体系与一些新型含能材料如二硝基酰铵(ADN)、硝仿肼(HNF)等相容性较差。因此,发展新的固化反应体系,对于提高固体推进剂的制造水平有着重要意义。
近年来,一些新型的固化反应方式引起了各国研究人员的重视。一是环氧基与胺基、酸酐等发生的开环加成反应[6];二是叠氮基与碳-碳双键、三键或碳-氮三键发生的1,3-偶极环加成反应[7]。上述反应固化时不受水分影响,具有宽温、宽环境适应性的优点。近期,本课题组开展了一系列腈氧化物固化剂与含碳-碳双键黏合剂的1,3-偶极环加成反应研究,证实该体系是一种能实现室温固化的新型固化体系。本文对上述固化体系的最新研究进展进行了详细介绍,并对固化效果和应用前景进行了对比评价,以期为新型固化体系的设计提供借鉴。
1 端环氧基-亲核基团固化体系
环氧基是一类具有—CH(O)CH—结构的官能团,由于三元环自身存在的高度张力,使得其可在较温和的条件下与胺基(伯胺、仲胺、酰胺)、羟基、巯基、羧酸、酸酐等亲核试剂发生开环加成反应,从而实现热固化甚至室温固化[8]。图1为环氧化合物与羧酸类化合物及伯胺类化合物发生固化反应的示意图。

图1 环氧化合物固化反应示意图Fig.1 The curing reaction of epoxides
HTPB是应用于固体推进剂体系中的一类重要黏合剂,通常采用异氰酸酯进行固化,对其端羟基的改性成为改变其固化方式的重要手段[9]。2011年,李娜等[10]以HTPB为引发剂,以BF3Et2O为催化剂,环氧氯丙烷为单体,发生阳离子开环聚合反应合成了两端为氯化聚醚的聚丁二烯(CTPB)。CTPB进一步在碱性条件下关环得到端环氧基聚丁二烯(ETPB)。ETPB的合成路线如图2所示。
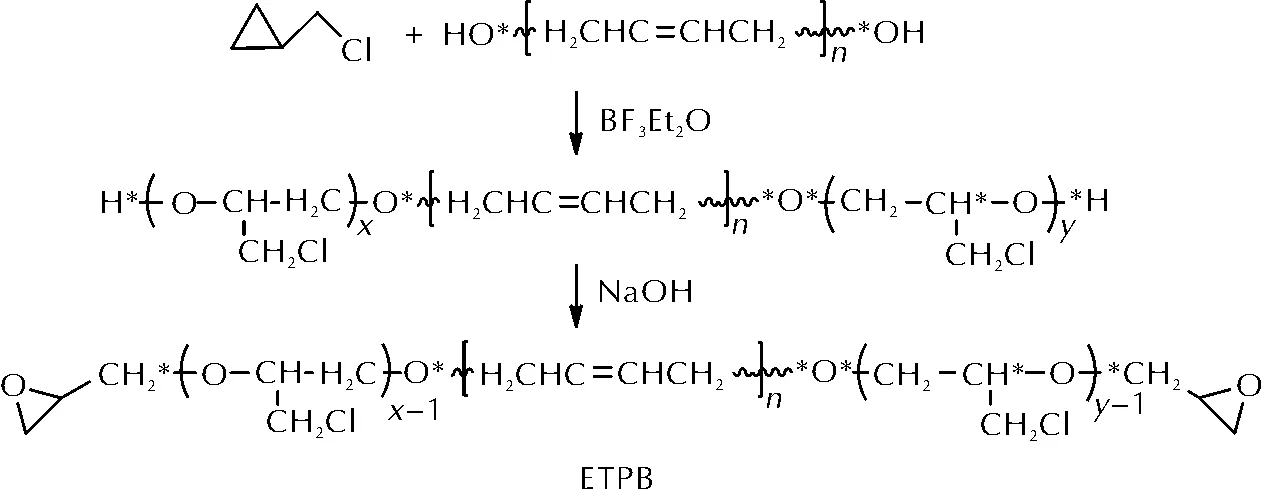
图2 端环氧基聚丁二烯(ETPB)的合成路线Fig.2 The synthetic route of ETPB
对ETPB固化反应的研究表明,采用2-甲基咪唑、甲基六氢邻苯二甲酸酐和聚酰胺650作为固化剂均可实现ETPB的固化。其中聚酰胺650在50℃时需4天将ETPB完全固化,固化后样品拉伸强度为4.5~5.1MPa,断裂伸长率为150%~180%,力学性能良好。
ETPB分子中的端环氧基团与惰性脂肪链相连,低温条件下难于与常规固化剂(胺类、酸酐类)发生固化反应。2012年,刘磊等[11]报道了一类钛酸酯偶联剂与ETPB的低温固化反应。钛酸酯偶联剂Tc-114的分子结构如图3所示。
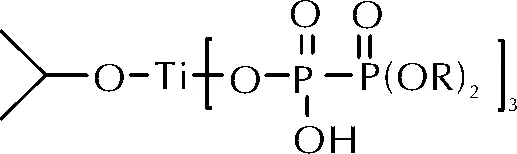
图3 钛酸酯偶联剂Tc-114的分子结构Fig.3 The molecular structure of Tc-114
固化动力学研究表明,该固化反应的表观活化能为40.76kJ/mol。当固化温度为30℃时,固化反应较慢,固化产物力学性能较差。当固化温度为50℃,TC-114质量分数为24%时,所得固化产物的交联密度最大,力学性能最佳。其拉伸强度为0.75MPa,断裂伸长率达110%。需要注意的是,ETPB随着环氧值增加,体系交联密度增大,其固化产物的拉伸强度随之增强,而断裂伸长率明显下降。
任晓婷等[12]以聚乙二醇或聚四氢呋喃-环氧乙烷共聚醚为原料,与金属钠或氢化钠反应得到聚醚二元醇钠,进而与环氧氯丙烷反应获得了多种端环氧基聚醚,如图4所示。
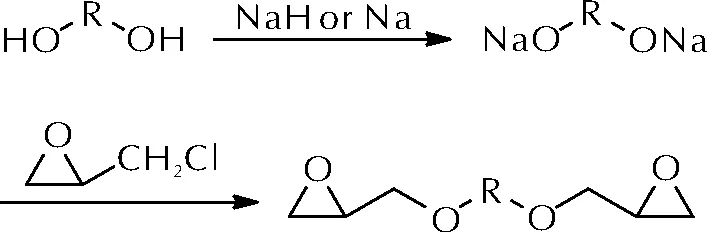
图4 环氧基聚醚黏合剂的合成路线Fig.4 The synthetic route of epoxy-teminated polyether
该系列环氧基聚醚黏合剂可实现一锅法制备,产物环氧值易控制,纯度不小于99%,收率在80%以上。进一步研究发现,上述端环氧基聚醚与聚酰胺650可在50℃、3天完成固化,所得胶片拉伸强度0.72~0.78MPa,断裂伸长率为66%~80%。
聚缩水甘油醚硝酸酯(PGN)是一种高能含能黏合剂,由于其聚氨酯网络的降解活化能较低,使用脂肪族异氰酸酯固化时产物在室温贮存后会发生严重降解。2017年,王伟等[13]在碱性条件下将PGN的端羟基与相邻的硝酸酯基发生脱酸成环反应转化为环氧基,合成了端环氧基PGN(e-PGN)。e-PGN的合成路线如图5所示。
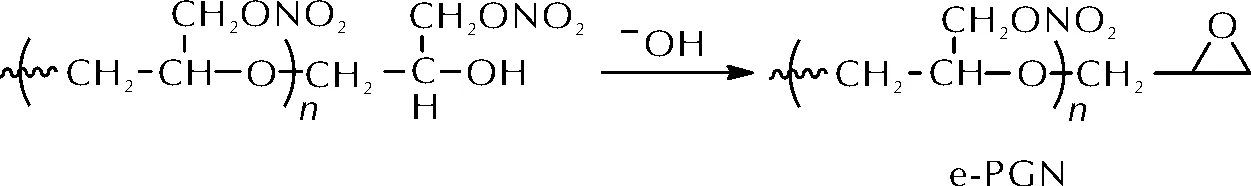
图5 端环氧基聚缩水甘油醚硝酸酯(e-PGN)的合成路线Fig.5 The synthetic route of e-PGN
e-PGN可在60~70 ℃下与多种固化剂发生固化反应,包括邻苯二甲酸酐(PA)、咪唑(IMD)、N-乙基乙二胺(NEED)和异佛尔酮二胺(IPDA)等。其中以PA为固化剂得到的固化产物力学性能最佳,拉伸强度为0.912MPa,断裂延伸率为354%,且固化胶片存放8个月后力学性能基本不变。
由上述可以看出,将多种端羟基黏合剂进行端环氧改性后,有效解决了异氰酸酯固化体系的水敏感问题,且保持了较好的力学性能。但目前此领域公开报道的文献不多,有待于更深入的研究。
2 叠氮基-炔基固化体系
叠氮基与炔基之间的1,3-偶极环加成反应又称Huisgen反应[14],可生成1,5-或1,4-取代的1,2,3-三唑化合物,其反应机制如图6所示。
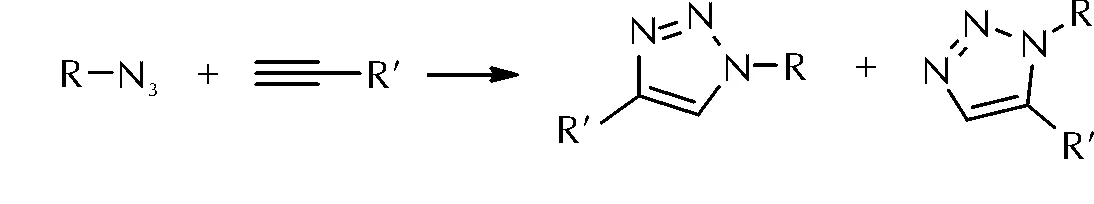
图6 叠氮基与炔基的1,3-偶极环加成反应Fig.6 1,3-Dipolar cycloaddition reaction of azide and alkynyl
此反应可应用于含能材料合成以及黏合剂的固化反应[15-16],具有以下几方面优点:(1)所需条件温和,对空气和原材料中的水分不敏感,适用范围较广;(2)所生成的三唑结构存在共轭效应,增强了结构的稳定性,有助于提高力学性能;(3)聚三唑结构本身带有一定的极性,故其弹性体与极性增塑剂相容性较好;(4)聚三唑结构含氮量丰富,生成焓较高,有助于提高固体推进剂的能量水平[17]。
2.1 叠氮黏合剂的炔基固化
聚叠氮缩水甘油醚(GAP)是由聚环氧氯丙烷与叠氮化钠合成的一种端羟基聚合物。其主链为聚醚结构,侧链含有大量的叠氮基团。GAP具有密度大、能量高、相容性好等优点,应用于固体推进剂中有助于提高能量水平、降低特征信号[18]。利用其侧链上叠氮基的活性,可实现GAP的非异氰酸酯条件固化。GAP及端炔基类固化剂的分子式见图7。
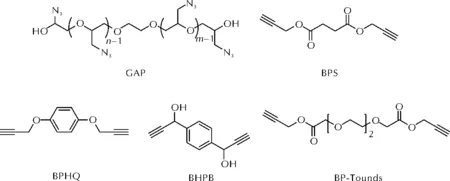
图7 GAP及端炔基固化剂BPS、BPHQ、BHPB和BP-Tounds的分子式Fig.7 The molecular structures of GAP, BPS, BPHQ, BHPB and BP-Tounds
2009年,Keicher等[19]使用不同官能度、不同分子量的GAP与丁二酸二丙炔醇酯(BPS)在45~65℃下实现了固化反应。实验结果显示,固化后的交联弹性体的玻璃化温度在-39~-34℃之间,断裂伸长率为50%~90%,弹性模量0.06~0.67MPa,拉伸强度0.05~0.32MPa,总体力学性能不高。Reshmi等[20]进一步通过DFT方法研究证实,GAP中的叠氮基与BPS中的炔丙基之间发生1,4-环加成反应的活化能(60kJ/mol)要高于1,5-环加成反应的活化能(58kJ/mol),故二者之间更倾向于发生1,5-环加成反应。Keicher等[21]还进一步将BPS的分子链延长,合成了新固化剂三氧杂十一烷二酸丙炔醇酯(BP-Tounds)。研究显示,虽然GAP-BP-Tounds交联弹性体强度较低,但由于其有效运动链段更长,导致相较于GAP-BPS交联弹性体,GAP-BP-Tounds交联弹性体的断裂延伸率更高,玻璃化温度更低。
使用端炔基化合物/异氰酸酯的双固化剂与GAP进行交联反应,有助于提高弹性体的力学性能[22]。Hagen等[23]使用对苯二丙炔酚/异佛尔酮二氰酸酯(BPHQ/IPDI) 固化体系在60℃下与GAP进行交联,所得弹性体断裂伸长率为72%~95%,拉伸强度0.60~0.85MPa,弹性模量0.71~1.28MPa。当加入BuNENA作为增塑剂时,其玻璃化温度可低至-57℃。Min等[24]发展了一类新型的双固化体系,以GAP为黏合剂,IPDI为扩链剂,多异氰酸酯(N100)、BPS或1, 4-双(1-羟基丙炔基)苯(BHPB)为固化剂,即同时存在叠氮/炔基和羟基/异氰酸酯的固化体系。该双固化体系的交联弹性体的拉伸强度不高,最高不超过0.588MPa,但是断裂伸长率很好,最高可达985%。说明由于双固化体系带来了不同的交联网络结构,对材料的力学性能产生了较大影响。Min等[25]还进一步将上述GAP基双固化体系应用于固体推进剂中,制备了GAP/AP/HMX等配方并进行了性能研究。相较于单独的三唑交联体系,双固化体系配方显示出更高的燃速和更低的压力指数。此外,实验证实该双固化体系GAP基推进剂与火箭的HTPB基内衬层有很好的黏附性,进一步提高了其应用价值。
国内王鑫等[26]同样利用1, 3-偶极环加成反应实现了GAP的固化。该课题组合成了三种炔基酯类固化剂,均苯三甲酸三炔丙酯(TPTM)、1, 6-六亚甲基二氨基甲酸甲基丁炔酯(HDPC)和1, 6-六亚甲基二氨基甲酸丁炔酯(DHD),在60~70℃下与GAP反应8~45h实现固化,其中TPTM固化活性最高。上述3种固化剂的分子式如图8所示。
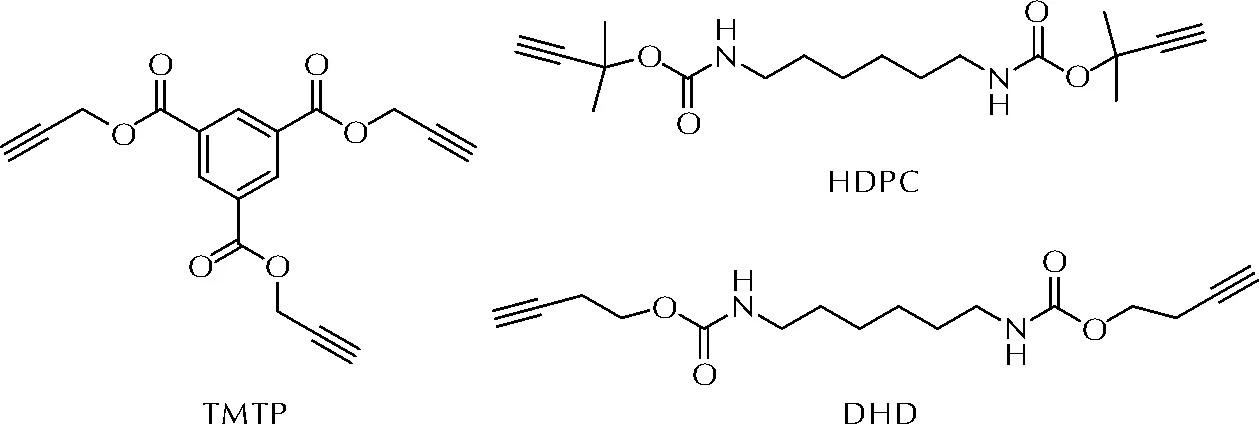
图8 端炔基固化剂TPTM、HDPC和DHD的分子式Fig.8 The molecular structures of TPTM, HDPC and DHD
力学性能研究显示,由于TPTM分子结构中存在刚性的苯环,导致在相同质量分数下,TPTM固化胶片的拉伸强度优于HDPC固化胶片和DHD固化胶片,而断裂延长率较低。另外,上述3种非异氰酸酯固化胶片内部都不存在气孔,进一步提高了燃烧性能的稳定性。
2016年,李辉等[27]通过环己烷二甲酸与丙炔醇反应合成了新型端炔基固化剂环己烷二甲酸丙炔醇酯(BPHA),制备方法如图9所示。
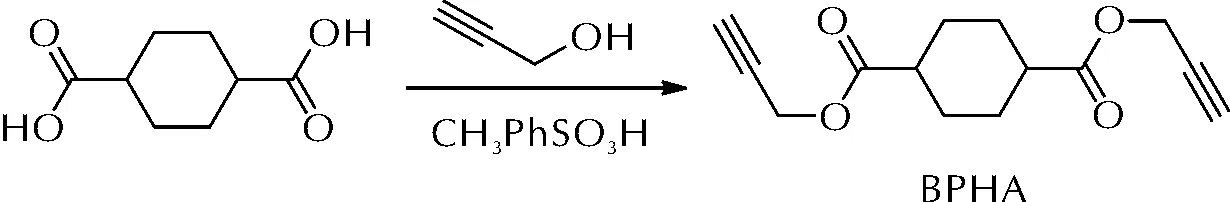
图9 BPHA的合成路线Fig.9 The synthetic route of BPHA
分别使用叠氮类黏合剂GAP和3-叠氮甲基-3-甲基氧丁环均聚物(PAMMO)与BPHA在60℃固化7天后制得相应的聚三唑弹性体。力学性能研究表明,由于PAMMO较GAP具有更有柔性的分子骨架,故基于PAMMO的弹性体较基于GAP的弹性体具有更大的拉伸强度和断裂延伸率。固化动力学研究显示,GAP与BPHA的反应速率要高于PAMMO。随着交联密度增大,两种弹性体的玻璃化温度均呈现升高趋势,其中基于PAMMO 的弹性体玻璃化温度升高速率更快。上述聚三唑弹性体起始分解温度均大于230℃,热稳定性好。
总体来说,由于GAP结构中大量甲基叠氮基团分布在分子链段的中间部位,固化时交联点随机分布且易于形成悬挂链,造成固化后形成的三维网络不完善,导致基于GAP的三唑交联弹性体力学性能较差。
2.2 端叠氮黏合剂的炔基固化
聚乙二醇(PEG)一方面拥有较低的玻璃化温度,另一方面其分子链上含有大量羟基,极性较高,与硝酸酯类增塑剂互溶性好,是NEPE推进剂中的重要组分[28]。冯增国等[29]采用硝硫混酸对不同相对分子质量的PEG进行硝化得到端硝酸酯基聚乙二醇(NTPEG),进而通过相转移催化叠氮化合成端叠氮基聚乙二醇(ATPEG)。ATPEG也可由端羟基聚乙二醇通过卤代、叠氮化反应制备而无需添加相转移催化剂[30]。ATPEG的两种合成路线见图10。
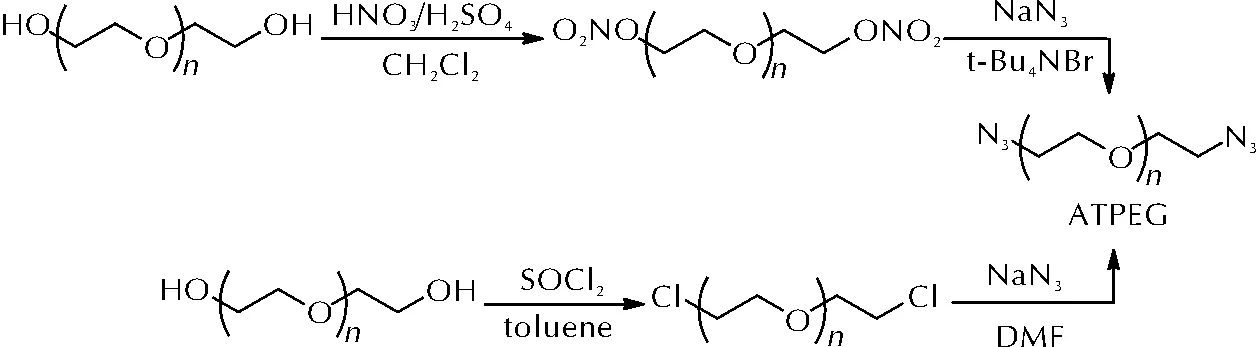
图10 ATPEG的两种合成路线Fig.10 Two synthetic routes of ATPEG
进一步研究显示,不同相对分子质量的ATPEG可以与N,N,N′,N′-四炔丙基乙二胺和1,1,1-三(炔丙氧甲基)丙烷固化得到交联弹性体。随着ATPEG 相对分子质量增大, 固化峰温越高,反应活化能随之下降[31]。此外,ATPEG 相对分子质量的增大有助于提高其在DMF和水中的溶胀比,采用三官能度的1,1,1-三(炔丙氧甲基)丙烷作为固化剂得到的胶片延伸率更佳[32]。
张倩等[33]将PET与2-氯乙基异腈酸酯反应后进一步叠氮化,合成了含有聚氨酯片段的液态端叠氮基环氧乙烷-四氢呋喃共聚醚(ATUPET),反应路线如图11所示。
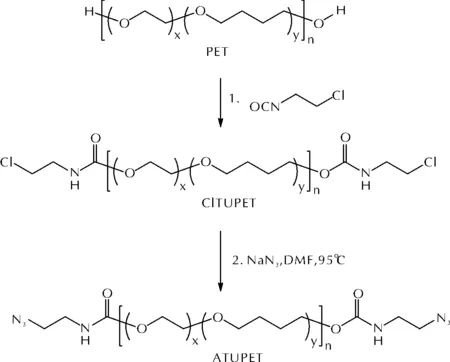
图11 ATUPET的合成路线Fig.11 The synthetic routes of ATUPET
ATUPET与三炔丙基胺在三氟甲磺酸四乙氰铜的催化下于50℃固化5天制得端位交联弹性体。随着R值的提高(0.7~1.3),弹性体的弹性模量(E)和拉伸强度(σb)均有明显提高(E=0.09~2.09MPa;σb=0.41~1.01MPa),而断裂伸长率则由1434.49%降至71.59%。该弹性体显示出优良的热稳定性,其热分解峰温接近400℃。
Lee等[34-36]报道了两种端叠氮基改性的聚己内酯(PCP 0260-N3和PCP 0310-N3),作为预聚物与多种亲偶极体端炔基固化剂进行固化反应研究。两种端叠氮基聚己内酯和端炔基固化剂的合成方法如图12所示。
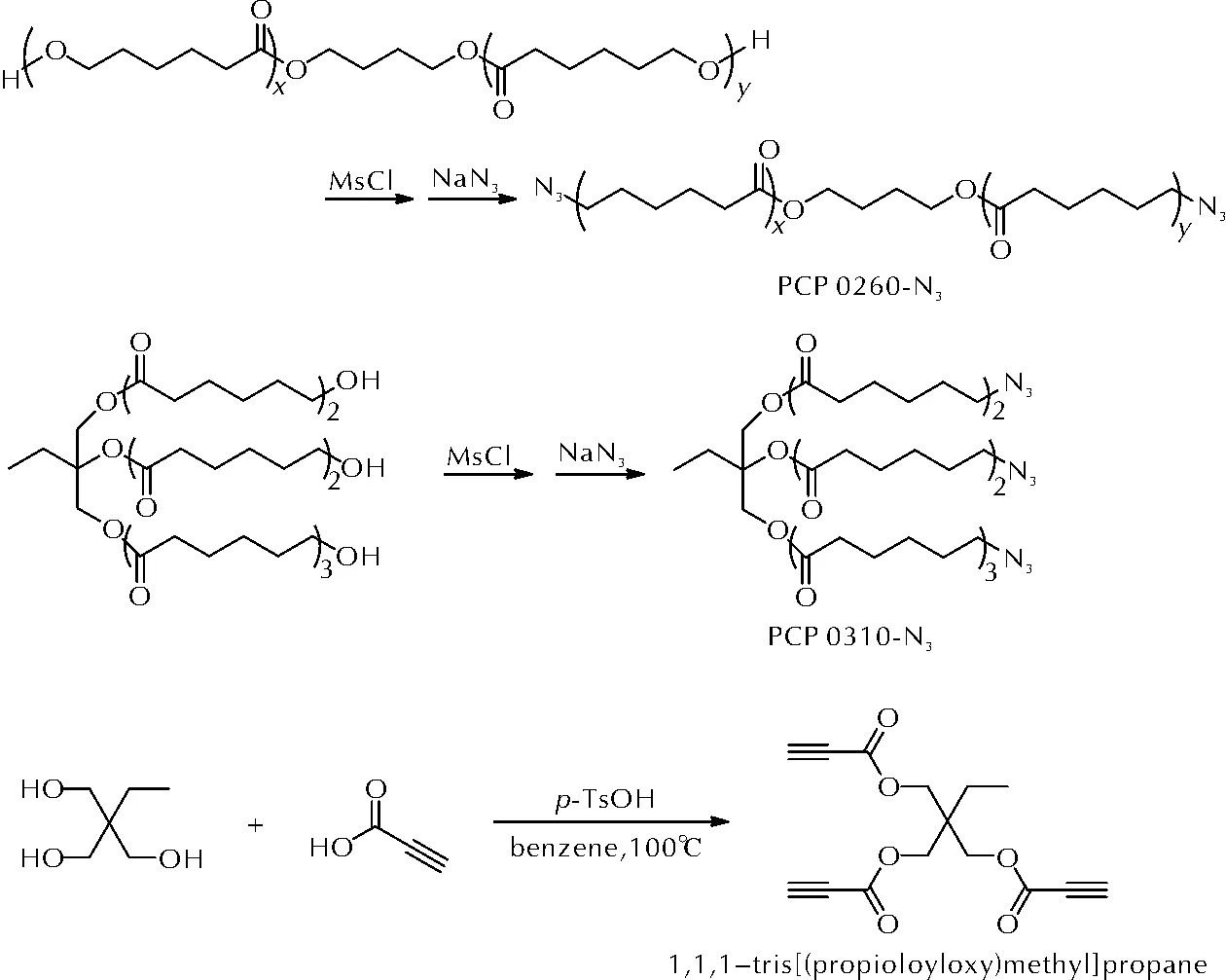
图12 PCP 0260-N3、PCP 0310-N3和1,1,1-三(丙炔酸甲酯基)丙烷的合成路线Fig.12 The synthetic routes of PCP 0260-N3, PCP 0310-N3 and 1,1,1-tris[(propioloyloxy)methyl]propane
综合比较之后可以看出,使用PCP 0260-N3作为预聚物,1,1,1-三(丙炔酸甲酯基)丙烷作为固化剂时,在52℃下反应7天可实现固化,且产物的力学性能较好。硝酸酯增塑剂的加入有助于提高产物的断裂延伸率并降低其玻璃化温度,但也会降低其拉伸强度。综合比较而言,加入质量分数为100%的增塑剂后所得产品的力学性能最好,拉伸强度可达2.467MPa,断裂伸长率达615.2%。此外,Wang等[37]和Katritzky等[38]还分别报道了多种端丙炔酸酯基聚合物与端叠氮基聚合物之间的固化反应,反应温度从室温至70℃,最高拉伸强度接近4.0MPa。
由上述可以看出,当叠氮基团位于黏合剂分子链段端位时,固化后交联点分布有序,可获得更为规整的三维立体网络结构,所得弹性体力学性能更为优良。
2.3 端炔基黏合剂的叠氮固化
聚乙二醇改性的另一种思路是将其端羟基转化为端炔基,再与多叠氮基化合物组成新的固化体系。杨荣杰等[39]以聚乙二醇400和丙炔溴为原料,合成了端炔基聚乙二醇(C≡PEG 400),合成路线如图13所示。
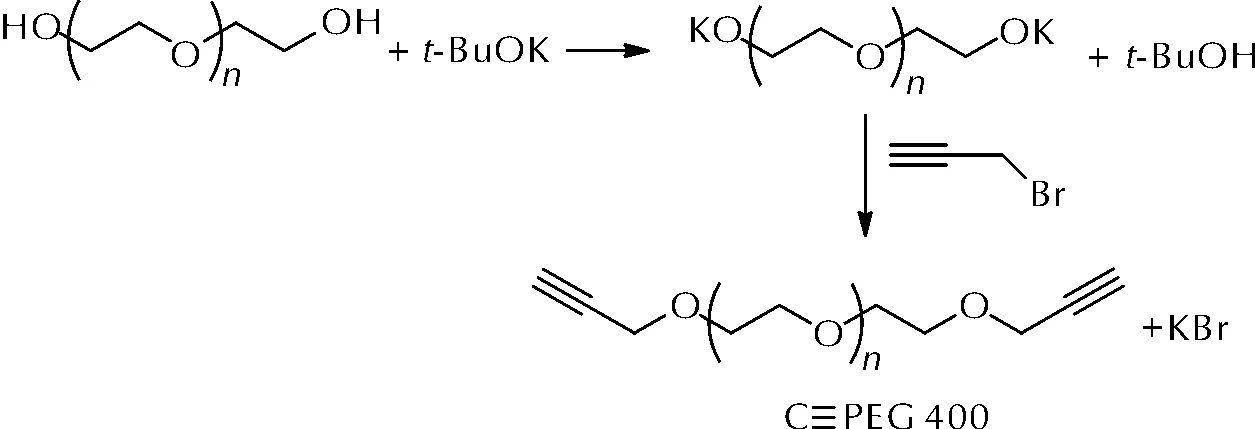
图13 C≡PEG 400的两种合成路线Fig.13 The synthetic route of C≡PEG 400
C≡PEG 400与多叠氮基固化剂反应结果表明,添加CuI做为催化剂有助于提高固化速率,而添加三醋酸甘油酯为增塑剂会延长固化时间。固化胶片平均拉伸强度为0.46MPa,平均应变率为20.6%,平均拉伸弹性模量为2.59MPa,拉伸强度不高。
环氧乙烷四氢呋喃共聚醚具有分子柔顺性好、玻璃化温度低、与硝酸酯增塑剂(NG、BTTN 等)的相容性好等多种优点,应用于固体推进剂中可改善其低温力学性能并降低感度[40]。杨荣杰等[41]合成了一种相对分子质量约4000的端炔基环氧乙烷-四氢呋喃共聚醚(C≡PET4000),在特定催化剂作用下可与官能度为3.8的叠氮化合物反应实现交联固化。结果显示端炔基聚醚-叠氮固化剂胶片的力学性能与端羟基聚醚-异氰酸酯基胶片相近,且叠氮基团与炔基基团摩尔比为1∶1时三唑胶片拉伸强度最大。
李辉等[42]通过Williamson 醚化法合成了端炔基聚丁二烯(PTPB)和端炔基环氧乙烷四氢呋喃嵌段共聚醚(PTPE),并与三(叠氮乙酸)三羟甲基丙烷酯(TMP-N3)进行了固化反应研究。PTPB、PTPE和TMP-N3的合成路线如图14所示。
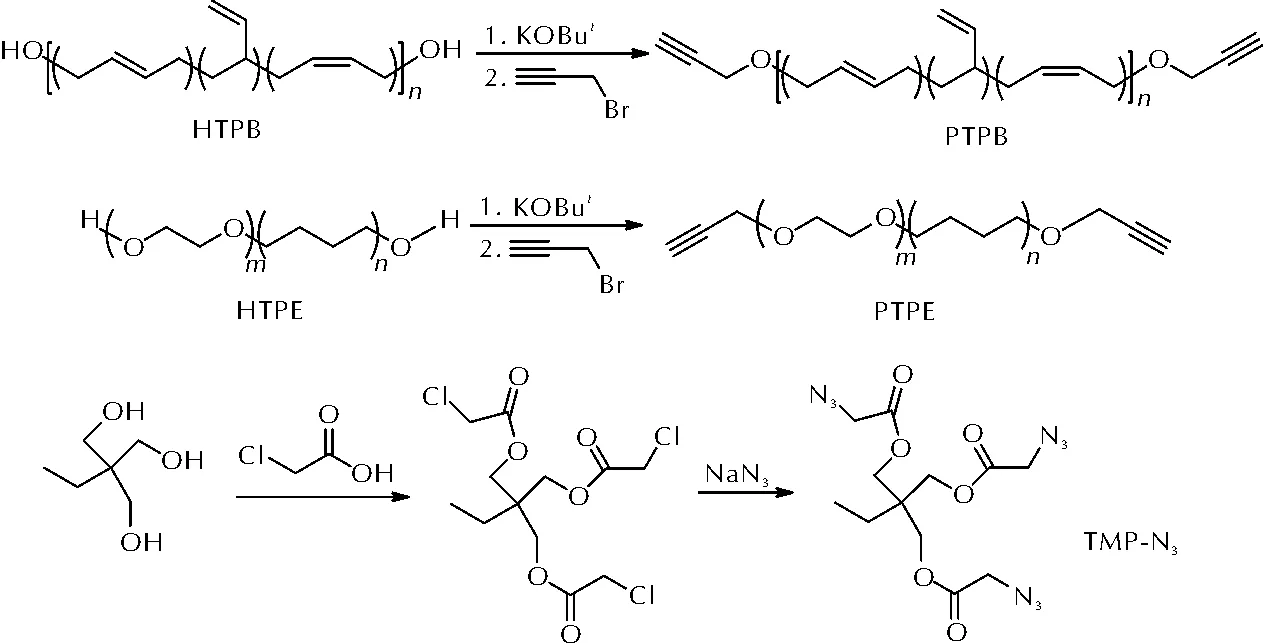
图14 PTPB、PTPE和TMP-N3的合成路线Fig.14 The synthetic route of PTPB, PTPE and TMP-N3
研究显示,炔基与羟基的摩尔数之比为1 时,基于PTPE 的聚三唑弹性体断裂延伸率更大(185.4%),而基于PTPB 的聚三唑弹性体拉伸强度更好(0.6MPa)。此外,SEM和AFM表征显示这两种聚三唑弹性体呈现出较为光滑的断面,说明内部结构均一,无气孔的形成。
由于硬段-硬段间的氢键作用和硬段微区作用,聚氨酯弹性体体现出优异的力学性能[43]。李辉等[44]构建了含氨基甲酸酯单元的端炔基黏合剂(PUPB),与TMP-N3进行了固化反应研究。结果显示,由于相邻链段之间酰胺官能团的氢键作用(如图15所示),有利于微相分离的形成,使得基于PUPB的聚三唑弹性体表现出较PTPB更为优异的力学性能(拉伸强度4.1MPa,断裂延伸率81.5%)。当加入葵二酸二异辛酯(DOS)作为增塑剂时,聚合物分子链段间的相互作用力进一步削弱,断裂延伸率可提升至293.3%。
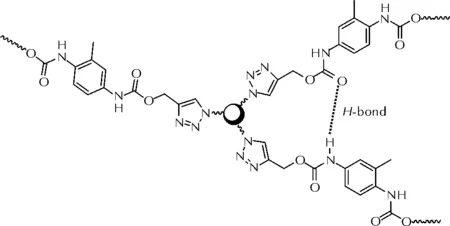
图15 PUPB聚三唑弹性体的分子间作用力Fig.15 The intramolecular interaction of PUPB-based trizaole-crosslinked polymers
HTPB也可以通过端基改性实现1,3-偶极环加成固化。Reshmi等[45]首先通过HTPB与TDI反应合成了端异氰酸酯预聚物(ITPB),然后通过异氰酸酯基与羟基的加成反应分别合成了端炔基预聚物(PrTPB)和端叠氮基预聚物(AzTPB),进而在60℃下制备了交联弹性体。预聚物的合成及固化反应方法如图16所示。PrTPB与AzTPB固化后测得的交联密度(2.86×10-4mol/cm3)高于其理论密度(5.01×10-5mol/cm3),说明端叠氮基与聚丁二烯骨架上的不饱和双键也同时发生了反应,生成了三唑-三唑啉网络结构。作者还制备了含铝粉-高氯酸铵-黏合剂配方的推进剂,并研究了其性能。结果表明,相较于传统的HTPB-TDI基推进剂,PrTPB-AzTPB基推进剂的黏度更低,力学性能提高了14%~22%,且燃速基本相同。
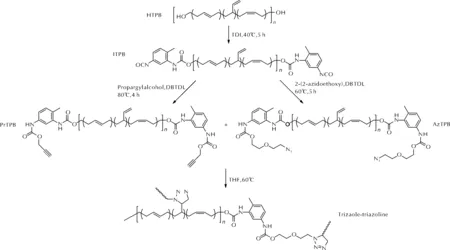
图16 PrTPB与AzTPB的合成路线及固化方法Fig.16 The synthetic route of PrTPB, AzTPB and curing method
总体来看,端炔基黏合剂经叠氮固化后弹性体的力学性能与端叠氮基黏合剂经炔基固化后弹性体的力学性能相似。端炔基改性的优势在于,端羟基黏合剂与叔丁醇钾/丙炔溴经一步反应即可实现官能团转化;而对于端叠氮基黏合剂,则需经氯化、叠氮化两步反应完成。
3 腈氧化物-烯基固化体系
腈氧化物是一类腈氧基(—CNO)直接与分子上的碳原子相连的有机化合物,其腈氧基官能团中含有高度极化的C—N键和C—O键[46]。Huisgen[14]将腈氧基同样归类为1,3-偶极体,能与含有亲偶极体的双键(三键)化合物发生[3+2]环加成反应,生成五元氮氧杂环。由于叠氮基团具有较强的爆炸性和毒性,加之需要亚铜离子催化,限制了其应用。随着人们对腈氧化物深入的研究,发现—CNO与炔、烯及氰基之间无需重金属催化即可发生高效、绿色的反应。对于含不饱和键的聚合物,可利用腈氧化物的1,3-偶极环加成反应来实现其交联[47]。图17为烯烃和腈氧化物发生1,3-偶极环加成反应得到异噁唑啉环五元氮氧杂环的过程。
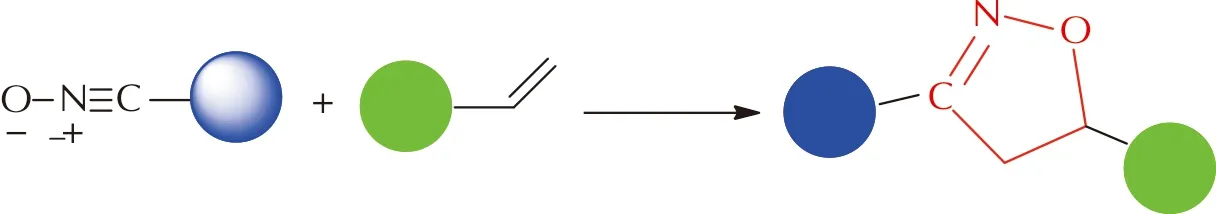
图17 烯烃和腈氧化物的1,3-偶极环加成反应Fig.17 The 1,3-dipolar cycloaddtion reaction of alkyne and nitrile oxide
但是,大多数芳香族和脂肪族腈氧化物在室温下都不能够稳定存在[48],容易发生二聚或异构化。因此,早先的研究以腈氧化物前驱体在使用时即时生成腈氧化物的方法解决其不稳定的问题。Huffman等[49]通过异氰酸酯和烷基硝酸酯反应制备出腈氧化物前驱体,在使用时加热分解生成腈氧化物,再与聚异戊二烯橡胶进行交联。2017年,樊亚勤等[50]将对苯二氯代醛肟和三乙胺反应,即时制备出高活性的对苯二腈氧化物(如图18所示),再与聚丁二烯(LPB)于27℃下反应制备出弹性体。研究发现,当对苯二氯代醛肟质量分数为10%时,制备弹性体的拉伸强度为0.35MPa,断裂伸长率为53%,玻璃化温度为-82.9℃。但是该方法制备腈氧化物前驱体时生成的铵盐会残留于体系中,从而对整体性能造成影响。
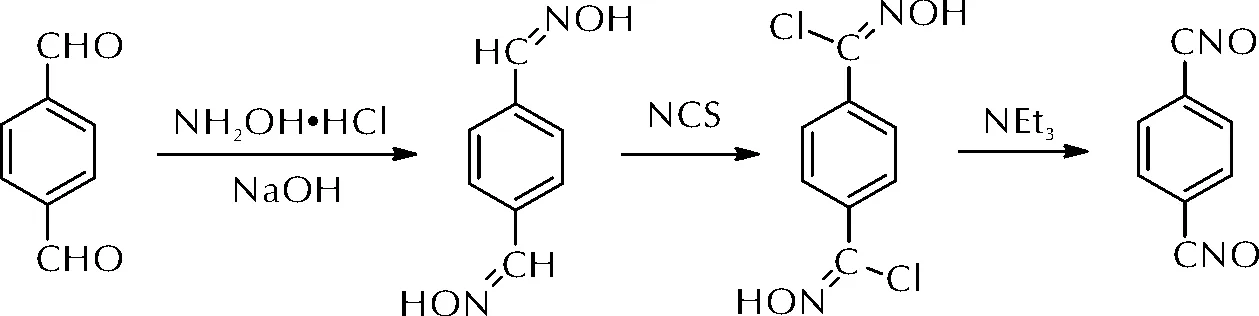
图18 对苯二腈氧化物的合成路线Fig.18 The synthetic route of terephthalonitrile oxide
因此,只有利用空间立体效应,在芳香族腈氧化物邻位引入位阻基团,才能使其稳定存在而避免二聚。早在20世纪90年代,Tsyganov等[51]发现,含有邻位位阻的苯基腈氧化物和联苯腈氧化物可在室温下稳定存在。2018年,王晓川等[52-53]分别以1,3,5-三甲苯和1,2,4,5-四甲苯为起始原料,经溴甲基化、氧化、缩合、脱氢氧化四步反应合成了2,4,6-三甲基间苯二腈氧化物(TINO)和1,2,4,5-四甲基对苯二腈氧化物(TTNO),上述两种腈氧化物的分子结构及TINO的合成路线如图19所示。
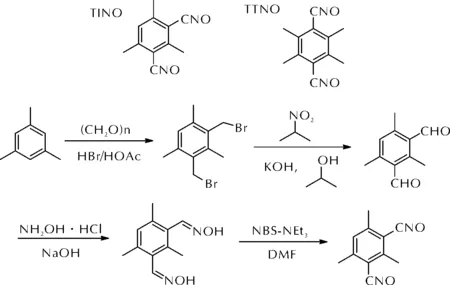
图19 TINO和TTNO的分子结构及TINO的合成路线Fig.19 The molecular structures of TINO and TTNO as well as the synthetic route of TINO
DSC测试表明,TINO和TTNO在100℃下无放热现象,其热分解放热峰温分别为133℃和179℃,说明上述固化剂具有较好的热稳定性。进一步固化实验显示,TINO和TTNO可分别与液体聚丁二烯(PB)和HTPB在25℃下、3天时间实现完全固化。其中对于TTNO与HTPB的固化胶片,随着固化剂含量(R值)的增大,弹性体的拉伸强度从0.135MPa增至0.416MPa,玻璃化转变温度从-73.08℃增至-66.70℃。
聚丁二烯类黏合剂经腈氧化物固化后拉伸强度偏低是因为双键过多且存在于分子链中,致使固化后形成的三维网络不完善,而将烯基置于聚合物端位则会改善此问题。莫洪昌等[54-55]利用PET中的羟基与烯丙基异氰酸酯中的异氰酸酯基团发生氨酯化加成反应,合成了一种含氨基甲酸酯基团的端烯基环氧乙烷-四氢呋喃共聚醚(AUPET),其结构如图20所示。
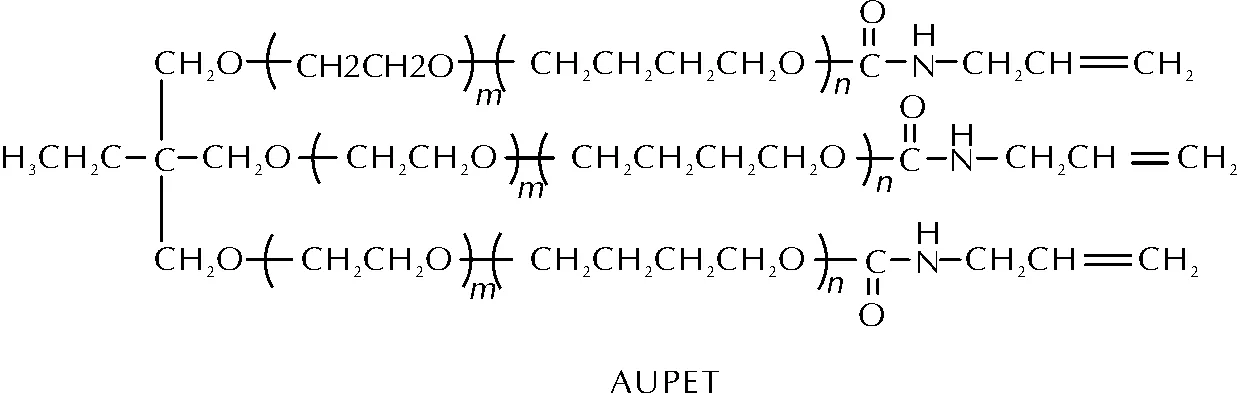
图20 AUPET的分子结构Fig.20 The molecular structure of AUPET
通过进一步研究发现,AUPET可与TINO在室温下平稳快速地实现固化,其凝胶时间仅为12h。反应生成的聚异噁唑啉弹性体在室温下的拉伸强度为1.75MPa,断裂伸长率为125%,明显优于TINO与HTPB固化产物的力学性能。
PNIMMO是一种采用3-硝酸酯甲基-3-甲基氧杂丁环(NIMMO)开环聚合后得到的一种端羟基聚合物。其主链结构为聚醚,每一个聚合单元含有一个硝酸酯基团,应用于PBX炸药或推进剂中可提高氧平衡和能量水平。通过在端位引入烯基的方法对其进一步改性,可实现其与腈氧化物之间的固化。王晓川等[56-57]以NIMMO为单体,使用1,4-丁二醇和三羟甲基丙烷为引发剂分别合成了与两官能度三嵌段端烯基PNIMMO(AGE-PNIMMO)和三官能度端烯基PNIMMO(AGE-T-PNIMMO)。分别将上述预聚物与TTNO于室温下固化5~7天后得到相应聚异噁唑啉弹性体。
通过对固化参数研究显示,R值为2.0时AGE-PNIMMO弹性体的拉伸强度最高,为0.60MPa;而AGE-T-PNIMMO最适宜的固化参数是R=1.0,其弹性体拉伸强度0.80MPa,断裂伸长率为150%。AGE-PNIMMO和AGE-PNIMMO弹性体的热分解峰温分别为216℃和202℃,热稳定性良好。
通过AGE聚合虽然可在PNIMMO端位引入双键,但引入数目不精确,导致固化后力学性能依然偏低。王晓川等[58]进一步将三官能度PNIMMO(T-PNIMMO)与3-异氰酸丙烯反应,制得三官能度AUT-PNIMMO含能黏合剂,其合成路线见图21。
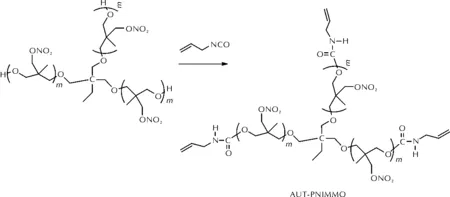
图21 AUT-PNIMMO的合成路线Fig.21 The synthetic route of AUT-PNIMMO

由上述可见,腈氧化物-烯基固化体系具有固化工艺简便、固化温度低、对水分不敏感等诸多优点。所制得弹性体的力学性能优异,热稳定性好,目前正在开展其在固体推进剂中的应用基础研究。
4 结束语
随着现代战争对战术火箭/导弹武器动力性能和推进效率要求的不断提高,迫切需要加快ADN、AlH3等新型高能材料在固体推进剂中的应用。然而,现役羟基/异氰酸酯固化体系由于存在毒性、水敏感性及相容性差等本质缺陷,难以保证新型高能固体推进剂加工和使用过程的安全性。近年来,各国研究人员陆续开发了端环氧基-亲核基团固化体系、叠氮基-炔基固化体系和腈氧化物-烯基固化体系等新型非异氰酸酯固化体系,合成出多种含能/非含能弹性体,重点研究了力学性能、热性能等基础性能。上述固化体系具有以下特点:
(1)端环氧基-亲核基团固化体系和叠氮基-炔基固化体系的固化温度较高,通常约50~70℃;腈氧化物-烯基固化体系则具有较高反应活性,可实现无催化条件下的室温固化,且固化过程无明显热效应,更有利于解决推进剂装药时因收缩应力导致的脱粘问题。
(2)基于聚醚、聚己内酯等惰性预聚物制备的交联弹性体整体力学性能较好,而基于GAP等含能预聚物的交联固化体系力学性能不佳。需要进一步通过结构改性等方法开发兼具优异力学性能和较高能量的弹性体。
建议在今后的研究工作中,一方面应进一步加强对上述固化体系的机理研究,探索分子构型、交联网络对固化条件和产品性能的影响规律,指导筛选出更多综合性能优异的聚合物结构并开展合成工作;另一方面应尽快开展新型非异氰酸酯固化体系在固体推进剂中的应用研究,探索适宜的配方组分和加工方式,加快新型高能固体推进剂的应用步伐。
