PN(CH2)nP配体/Cr(Ⅲ)/DMAO/AlEt3催化乙烯齐聚
2021-09-04常琪琪范昊男王亚婷
常琪琪,王 讯,范昊男,王亚婷,姜 涛
(天津科技大学 化工与材料学院,天津 300457)
线性α-烯烃作为有机化工原料或者中间体,不仅是聚乙烯生产中的重要共聚单体(C4~C8),还可用于生产增塑剂(C6~C8)、润滑油品添加剂(C8~C12)以及洗涤剂(C16~C18)等[1-3]。近年来,全球对线性α-烯烃的需求量逐年增加[4],预计到2022年的需求量可达到6.50 Mt/a。由于中国线性α-烯烃产业起步较晚,目前仍需大量进口国外的高质量乙烯齐聚产品[5],这严重限制了中国高性能聚烯烃和高端润滑油产业的发展,成了“卡脖子”的技术难题[6-7]。由于链增长机制,使用传统非选择性乙烯齐聚技术的产物呈现S-F分布,如需要单一碳数的线性α-烯烃,则需要通过高精度精馏分离实现[8]。乙烯选择性齐聚可以高选择性地生产单一线性α-烯烃,该技术具有更高的原子利用率和产品价值。因此开发乙烯选择性齐聚生产C6~C10线性α-烯烃技术意义重大。
配体是乙烯选择性齐聚技术的关键,研究人员对此做过很多研究探索。2004年南非Sasol公司开发了第一种可用于催化乙烯选择性四聚的双膦胺(PNP)配体[9]。2012年Shaikh等[10]在上述PNP配体结构的基础上,设计合成了不对称的PNP型配体,其与CrCl3(THF)3络合,在MAO助催化作用下催化乙烯齐聚,催化活性达到2.3×106g/(mol Cr·h),乙烯齐聚产物呈宽分布;在DMAO/AlEt3助催化作用下催化乙烯齐聚,催化活性达到5.3×105g/(mol Cr·h),乙烯齐聚产物以己烯和辛烯为主。2017年笔者所在课题组Zhang等[11]合成了PNSiP型不对称配体,乙烯齐聚催化活性可达16.8×106g/(mol Cr·h),对1-己烯和1-辛烯的选择性分别为75%、25%。2018年Alam等[12]报道了硅化二磷胺(PNSiCP型配体)铬(Ⅲ)基体系,该体系催化乙烯齐聚活性为2.3×106g/(mol Cr·h),对1-己烯和1-辛烯的选择性分别达到16%、83%。在已报道的体系中,不对称PNP型配体铬(Ⅲ)基体系在乙烯齐聚催化中具有较好的稳定性,表现出了较高的催化活性和良好的1-己烯与1-辛烯选择性,其中,配体结构中N取代基以及配体骨架结构对催化剂性能有重要影响,故而新型不对称PNP型配体的研究对提高乙烯齐聚催化活性及催化体系的热稳定性有重要参考意义。
笔者设计合成了带有双齿膦乙基或双齿膦正丙基的不对称PN(CH2)nP配体,与CrCl3(THF)3、DMAO/AlEt3组成催化体系用于催化乙烯齐聚,考察了配体结构、反应温度、反应压力、Al/Cr摩尔比以及反应时间对该催化体系活性和产物选择性的影响。
1 实验部分
1.1 试剂和原料
二苯基氯化膦(质量分数98%)、二苯基膦(质量分数98%)、2-甲氨基乙醇(质量分数98%)、2-(异丙氨基)乙醇(质量分数99%)、3-(异丙氨基)丙醇(质量分数98%)、叔丁醇钾(质量分数98%)、氯化亚砜(质量分数98%)、无水硫酸钠(质量分数99%),均购自百灵威科技有限公司;三乙胺、三氯甲烷、四氢呋喃、甲基环己烷、正庚烷、乙醚,均为分析纯,购自萨恩化学技术有限公司,经分子筛干燥,金属钠回流后使用;甲基铝氧烷(MAO,1.4 mol/L 的甲苯溶液)、三乙基铝(AlEt3)(质量分数99.9%),均购自Albemarle公司;脱除挥发性组分的甲基铝氧烷(Dried methylaluminoxane,DMAO):MAO通过真空干燥除去三甲基铝(TMA)后得到的白色粉末;高纯氮气(质量分数99.2%),购自天津飞林气体有限公司;聚合级乙烯(质量分数99.9%),购自天津赛美特特种气体有限公司。
1.2 PN(CH2)nP配体的合成及表征
1.2.1 PN(CH2)nP配体的合成
PN(CH2)nP配体合成过程如图1所示。
配体L1、L2、L3中的R分别为甲基、异丙基、异丙基,n分别为2、2、3。
按照图1中PN(CH2)nP配体合成路线分别制备配体L1、L2、L3。
(1)氮气保护下,在3个三口圆底烧瓶中,分别加入3.75 g 2-甲氨基乙醇、5.16 g 2-(异丙氨基)乙醇、5.86 g 2-(异丙氨基)丙醇各溶于25 mL三氯甲烷中,三口烧瓶置于0 ℃冰水浴中恒温,持续搅拌,缓慢加入7.73 g氯化亚砜。滴加完成后,均在60 ℃回流反应3 h,在真空下减压蒸馏除去溶剂得到橙色晶体,用丙酮洗涤3次,分别得到白色晶体5.91 g HN(CH3)(CH2)2Cl·HCl、6.65 g HN(CH)(CH3)2(CH2)2Cl·HCl、6.87 g HN(CH)(CH3)2(CH2)3Cl·HCl。
(2)在3个三口圆底烧瓶中,分别加入4.38 g叔丁醇钾溶于10 mL四氢呋喃中,均加入2.90 g二苯基磷,室温下搅拌30 min。分别加入2.96 g HN(CH3)(CH2)2Cl·HCl、3.32 g HN(CH)(CH3)2(CH2)2Cl·HCl、3.43 g HN(CH)(CH3)2(CH2)3Cl·HCl,均在85 ℃下加热回流12 h,真空下减压蒸馏除去四氢呋喃溶剂。分别在残渣中加入5 mL质量分数为38%的盐酸溶液,搅拌直至出现白色盐状物,各用15 mL乙醚萃取3次,各在水相中加入5 mL质量分数为20%的氢氧化钠溶液洗涤,均用15 mL乙醚萃取3次,将有机层用无水硫酸钠进行干燥,过滤分别得到淡黄色油状物2.30 g HN(CH3)(CH2)2P(C6H5)2、3.24 g HN(CH)(CH3)2(CH2)2P(C6H5)2、3.75 g HN(CH)(CH3)2(CH2)3P(C6H5)2。
(3)在氮气保护下,在第二步得到的3种中间产物中分别加入0.01 g三乙胺,边搅拌边缓慢滴加2.21 g二苯基氯化膦,室温下搅拌12 h。用砂芯漏斗过滤除去三乙胺盐酸盐沉淀,滤液经浓缩、重结晶,分别得到白色粉末状产物3.64 g (C6H5)2PN(CH3)(CH2)2P(C6H5)2(L1)、淡黄色油状物2.23 g (C6H5)2PN(CH)(CH3)2(CH2)2P(C6H5)2(L2)、淡黄色油状物3.83 g (C6H5)2PN(CH)(CH3)2(CH2)3P(C6H5)2(L3),收率分别为90%、41%、62%。

L1:R=Methyl,n=1;L2:R=Isopropyl,n=2;L3:R=Isopropyl,n=3图1 PN(CH2)nP配体的合成过程及结构Fig.1 Synthesis routine and structures of the PN(CH2)nP ligand
1.2.2 PN(CH2)nP配体的表征
采用德国Bruker公司所产的ACANCEⅢ400M型核磁共振谱仪对配体结构进行表征。
配体L1的核磁表征:1H NMR(400 MHz,CDCl3,298 K)δ2.25~2.29(m,2H),2.51-2.52(d,3H),3.18~3.21(m,2H),7.30~7.38(m,20H)。13C NMR(400 MHz,CDCl3,298 K)δ 28.15~28.35,37.03,53.20~53.73,128.11~128.56,131.91,132.10~132.78,138.28,138.40,139.04,139.19。31P NMR(400 MHz,CDCl3,298 K)δ-21.01(s),63.28(s)。
配体L2的核磁表征:1H NMR(400 MHz,CDCl3,298 K)δ1.12~1.13(d,6H),1.63~1.67(m,2H),2.98~3.04(m,2H),3.25~3.31(m,1H),7.27~7.39(m,20H)。13C NMR(400 MHz,CDCl3,298 K)δ23.46,23.55,29.35,29.51,46.30,46.58,51.73,51.94,65.84,128.09~128.42,132.04~132.64,138.08,138.20,139.94,140.08。31P NMR(400 MHz,CDCl3,298 K)δ-20.04(s),45.61(s)。
配体L3的核磁表征:1H NMR(400 MHz,CDCl3,298 K)δ1.12-1.13(d,6H),1.29~1.35(m,2H),1.63~1.67(m,2H),2.98~3.04(m,2H),3.25~3.30(m,1H),7.27~7.39(m,20H)。13C NMR(400 MHz,CDCl3,298 K)δ23.38,23.47,25.34,25.46,26.88,27.06,50.39,50.54,51.33,51.54,128.02~128.41,132.08~132.69,138.75,138.8,140.33,140.47。31P NMR(400 MHz,CDCl3,298 K)δ-16.01(s),46.21(s)。
1.3 乙烯齐聚反应及产物分析
1.3.1 乙烯齐聚反应
乙烯齐聚反应在100 mL高压釜中进行。高压釜经高纯氮气和乙烯各置换3次,用循环水将温度调整到100 ℃后,依次加入20 mL甲基环己烷、助催化剂DMAO/AlEt3和配体,搅拌2 min后迅速加入铬化合物催化剂,调节高压釜至预定的温度和乙烯压力,乙烯齐聚反应进行30 min后用冰冻乙醇淬灭反应,然后降温、卸压。
1.3.2 乙烯齐聚产物分析
采用美国安捷伦公司生产的Agilent technologies 7890A型气相色谱仪,分析乙烯齐聚液相产物,以正庚烷为内标物,分别计算乙烯齐聚催化体系的催化活性和各乙烯齐聚液相产物的选择性,计算公式如式(1)、(2)所示。
(1)
(2)
式中,a为催化活性,g/(mol Cr·h)[13];si为线性α-烯烃的选择性,%;mtotal为反应产物总质量,g;mα为反应产物中线性α-烯烃质量,g;n为催化剂活性金属中心物质的量,mol;t为反应时间,h。
固相产物经质量分数为10%的酸化乙醇溶液洗涤,60 ℃真空干燥,称重[8,10,14-18]。
2 结果与讨论
2.1 反应温度对乙烯齐聚催化体系性能的影响
以甲基环己烷为溶剂,考察了反应温度对PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系性能的影响,结果见表1。由表1可看出,随反应温度的升高,PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系的催化活性呈先升高后降低的趋势。这是因为:随着反应温度升高,反应体系内分子的平均动能增大,齐聚反应链增长速率和链转移速率加快[19],从而使催化活性提高;但是,乙烯在溶剂中的溶解度随温度升高逐渐下降,且催化剂在120 ℃的高温下容易失活,从而导致活性降低。当反应压力为4 MPa时,配体L1和L3所组成催化体系的催化活性在80 ℃时达到最大值,分别为5.91×106、1.95×106g/(mol Cr·h),配体L2在100 ℃时催化活性达到最大值17.1×106g/(mol Cr·h)。说明L2组成的催化体系具有更好的高温稳定性。随着反应温度的升高,乙烯齐聚产物C10+选择性增加。以配体L2为例,当反应温度达到80 ℃时,1-己烯和1-辛烯总选择性最高,可达62.13%,可能因为此条件下催化体系中氢消除速率比扩散速率快,从而使乙烯齐聚产物分布以1-己烯和1-辛烯为主,C10+产物总选择性为35.66%;继续升温至120 ℃,催化剂可能会在高温下部分失活,此条件下催化体系中氢扩散速率比氢消除速率快,1-己烯和1-辛烯总选择性降低到35.35%,C10+产物总选择性增至60.83%,出现产物分布较宽的趋势[20]。这种当反应温度过高时α-烯烃产物选择性降低的现象,与报道的均相铬催化剂的催化反应结果一致[21-22]。
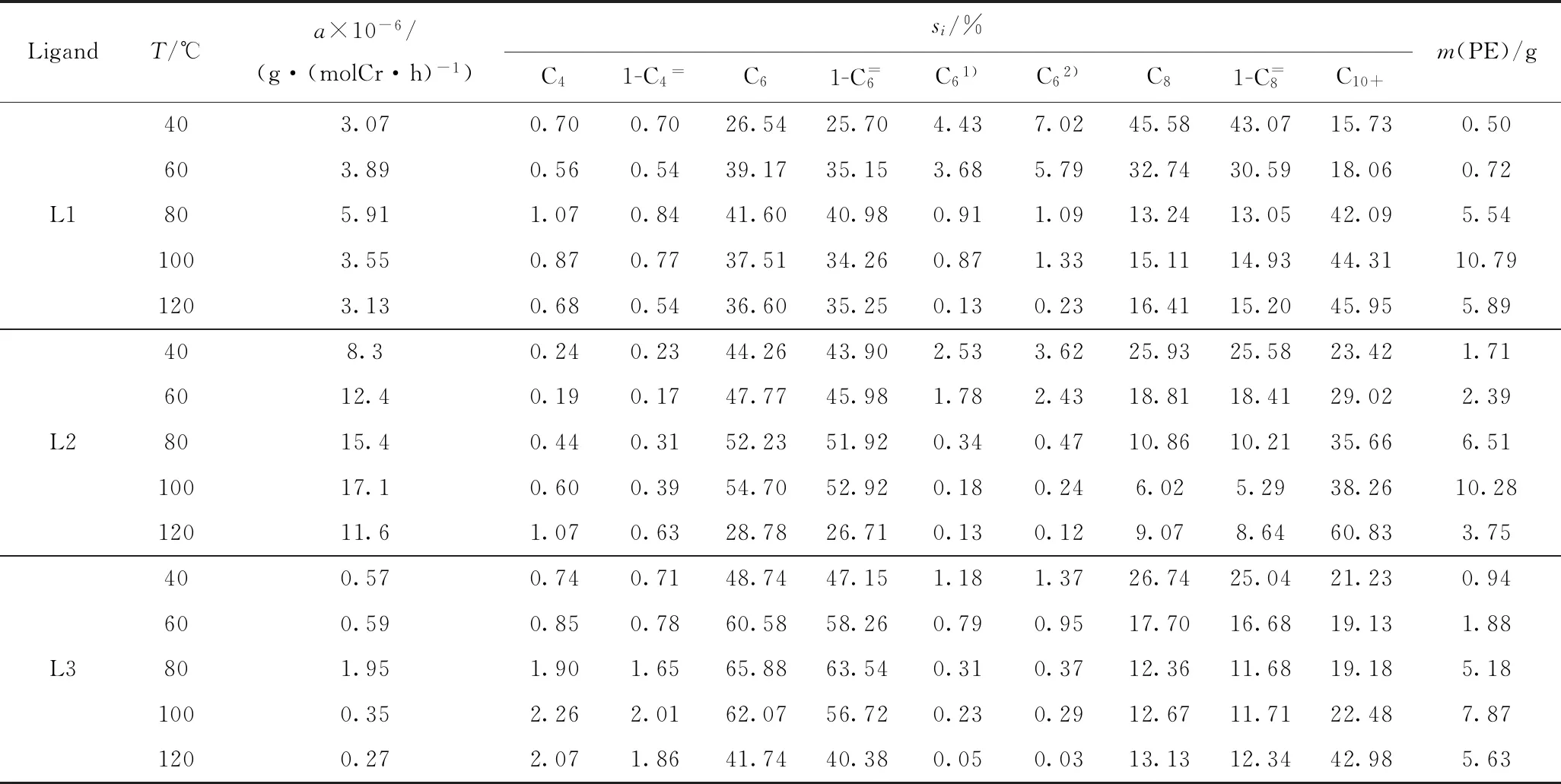
表1 反应温度对乙烯齐聚催化体系性能的影响Table 1 Effects of reaction temperature on the performance of ethylene oligomerization catalysis system
2.2 反应压力对乙烯齐聚催化体系性能的影响
以甲基环己烷为溶剂,考察了反应压力对PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系性能的影响,结果列于表2。由表2可以看出,随着反应压力的升高,PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系的催化活性呈上升的趋势。这是因为,在反应动力学上,随着反应压力升高,气相乙烯分子向溶剂扩散速率加快[20],反应体系中乙烯单体的浓度增大,乙烯分子向活性中心进攻参与配位插入的几率增大,表现为催化体系的催化活性提高。随着压力升高,乙烯齐聚产物C10+选择性增加。以配体L2所在的催化体系为例,当反应压力为1.0 MPa时,1-己烯和1-辛烯的总选择性高达93.43%,几乎没有C10+产物;当反应压力升高至4.0 MPa时,1-己烯和1-辛烯的总选择性仅为58.21%,C10+产物总选择性增至38.26%。分析其原因,可能是反应压力升高,对催化剂的催化行为产生了显著的影响。在较高反应压力下乙烯齐聚催化体系中氢扩散速率比氢消除速率快,从而导致C10+产物选择性增加,这种在反应压力升高时产物选择性分布较宽的现象,与报道的均相铬催化剂的催化反应结果是一致的[12,14,21-22]。

表2 反应压力对乙烯齐聚催化体系性能的影响Table 2 Effects of reaction pressure on the performance of ethylene oligomerization catalysis system
2.3 Al/Cr摩尔比对乙烯齐聚催化体系性能的影响
以甲基环己烷为溶剂,考察了不同Al/Cr摩尔比对PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系性能的影响,结果见表3。由表3可知,随着助催化剂DMAO/AlEt3摩尔浓度增加,PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系的催化活性不断增加[23-24]。这是因为在乙烯齐聚反应体系中,当Al/Cr摩尔比为300时,助催化剂DMAO/AlEt3摩尔浓度较低,大部分用于除去反应体系中水、氧等杂质,剩余的量不足与主催化剂和配体作用形成活性物种;而当Al/Cr摩尔比为700时,助催化剂DMAO/AlEt3摩尔浓度较高,可能使催化剂活性中心被过度还原生成非选择性齐聚物,或者催化体系催化活性增加后α-烯烃发生共聚,1-己烯和1-辛烯选择性下降,C10+产物选择性有所增加。因此,当3种催化体系Al/Cr摩尔比为500时为最佳,乙烯齐聚催化活性较高,且1-己烯和1-辛烯的总选择性最好,分别为89.42%、93.43%、92.94%。

表3 Al/Cr摩尔比对乙烯齐聚催化体系性能的影响Table 3 Effects of Al/Cr molar ratio on the performance of ethylene oligomerization catalysis system
2.4 反应时间对乙烯齐聚催化体系性能的影响
以甲基环己烷为溶剂,考察了反应时间对PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系性能的影响,结果见表4。由表4可知,对于L1/CrCl3(THF)3/DMAO/AlEt3催化体系来说,最佳反应时间为15 min,这表明该乙烯齐聚反应体系可以在较短的反应时间被完全活化。L2(L3)/CrCl3(THF)3/DMAO/AlEt3催化体系最佳反应时间均为30 min,增加反应时间会降低该催化体系的催化活性,对1-己烯和1-辛烯的选择性基本没有影响。

表4 反应时间对乙烯齐聚催化体系性能的影响Table 4 Effects of reaction time on the performance of ethylene oligomerization catalysis system
2.5 配体结构对乙烯齐聚催化体系性能的影响
以甲基环己烷为溶剂,考察了配体结构对PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系性能的影响,结果见表5。L1/CrCl3(THF)3/DMAO/AlEt3体系催化乙烯齐聚产物中,1-己烯和1-辛烯的总选择性为89.42%,活性为1.11×106g/(mol Cr·h)。将与P相邻的N上的取代基甲基(L1)换成异丙基(L2)后,其组成的催化体系乙烯齐聚产物中1-己烯和1-辛烯的总选择性可达到93.43%,且高压条件下其催化活性较L1提高了10倍,可达到1.71×107g/(mol Cr·h)。这可能是由于配体更换取代基后,L2中心N原子的空间位阻增大[25],从而使催化性能发生显著变化。将L2骨架中乙基换为正丙基得到L3,其组成的催化体系乙烯齐聚产物中1-己烯和1-辛烯的总选择性为92.94%,但活性降低约为L2的1/10,为0.26×106g/(mol Cr·h)。其原因可能是亚甲基的加入使P1-Cr-P2咬合角变大,不利于骨架取代基与催化中心之间的相互作用,较大的P1-Cr-P2咬合角也可能是配体与CrCl3(THF)3之间弱配位的原因之一,从而降低催化活性。

表5 配体结构对乙烯齐聚催化体系性能的影响Table 5 Effects of ligand structure on the performance of ethylene oligomerization catalysis system
3 结 论
(1)当反应温度为100 ℃、反应压力为4.0 MPa时,L2/CrCl3(THF)3/DMAO/AlEt3体系催化乙烯齐聚的活性可达到1.71×107g/(mol Cr·h),该催化体系具有较高的热稳定性。
(2)在不同压力下,3种PN(CH2)nP/CrCl3(THF)3/DMAO/AlEt3催化体系产物分布差异很大。当反应压力为1.0 MPa时,3种催化体系产物以1-己烯和1-辛烯为主;当反应压力为4.0 MPa时,3种催化体系1-己烯和1-辛烯产物选择性降低,C10+产物选择性有所增加。
(3)配体结构对催化性能有较大影响。当配体L1中引入空间位阻较大的取代基团异丙基(L2)时,乙烯齐聚活性显著提高,且热稳定性好;将L2骨架中乙基换为正丙基得到L3,其活性和稳定性均降低,可能是由于P1-Cr-P2咬合角变大造成的。
