5,15-二(二茂铁基)-卟啉酞菁钇的结构和振动光谱的密度泛函理论研究
2021-08-16陈玉锋
姜 力,周 启,陈玉锋,蔡 雪
(牡丹江师范学院化学化工学院,牡丹江市 157000)
1 引 言
卟啉和酞菁作为最常见也是最重要的四吡咯衍生物,由于其具有比较独特的光、电、磁性质,引起了科学家们的广泛关注[1,2].它们是一类具有18π电子的多吡咯共轭化合物,可以同多种稀土金属配位构成双层或三层的三明治型配合物[3],在这些分子中,彼此之间被拉到很近距离的两个或三个共轭π体系之间相互作用,再加上稀土金属中心固有的性质[4],使这些配合物表现出三明治型结构独特的性质,使它们在分子电子学,分子信息储存,气体传感,电致变色和有机场效应晶体管等方面具有广泛应用[5-8].
密度泛函理论(density functional theory,DFT)与实验数据相结合的方法,常被用来研究分子的几何构型和振动光谱[9-11],由于B3LYP方法对计算资源要求不高并且具有较高的精确度,所以该方法在理论计算上应用较广泛.Seog Woo等人研究合成了5-二茂铁-15-苯基-2,8,12,18-四乙基-3,7,13,17-四甲基卟啉(1),并分别与Zn(OAc)2·2H2O和Mncl2·4H2O金属化反应得到Zn(1)、Mncl(1).在0.29、0.22、0.37V下,(1)、Zn(1)、Mncl(1)的循环伏安图显示了二茂铁的可逆单电子氧化过程[12].Boyd等人合成了5,15-二(二茂铁)-2,8,12,18-四叔丁基-3,7,13,17-四甲基卟啉并得到了其镍的配合物,发现该配合物呈syn构型分布,在两个二茂铁基团之间存在电子耦合[13].尽管有四吡咯配合物体系中引入二茂铁基团的很多例子被报道过,但在其三明治型配合物结构框架中引入二茂铁基团并同时混杂卟啉与酞菁的例子却很少.本文采用混杂的B3LYP密度泛函方法[14,15],在Lanl2dz基组水平上,计算5,15-二(二茂铁基)-卟啉酞菁钇分子[Por(Fc)2]Y(Pc)的红外光谱,与实验所得的红外光谱图进行了比对,利用Gauss-View软件对5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的简正振动模式进行了指认归属[16],分析研究了5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的分子表面静电势.
2 理论计算和实验
值得提及的是,由于[Por(Fc)2]Y(Pc)分子中既包含稀土金属钇又含有过渡金属铁,再加上整个分子体系的所有原子共有129个之多,对于这样复杂的分子结构,一些小的基组如被大家熟知并被广泛使用的6-31G(d)基组不能被用来计算.因此在本文工作中选择了Lanl2dz基组[17],在计算过程的能量最小化环节采用了使用冗余内坐标的Berny运算法则,全部计算过程采用默认的收敛标准,在前面步骤产生的能量最小化结构基础上进行了正则坐标分析[18,19],计算所得的振动光谱频率用0.9614校正系数进行校正[20].理论计算采用Gaussian09量子化学程序包[21],分子构型用GaussView 6.0构造,利用Gaussian 09程序包,在B3LYP/Lanl2dz水平上对5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的几何结构进行优化和频率计算,简正振动模式分析指认归属采用GaussView软件完成.
5,15-二(二茂铁基)-卟啉酞菁钇分子[Por(Fc)2]Y(Pc)的红外光谱(IR)采集使用BIORAD FIS-165分析仪,波数范围为4000-400 cm-1,分辨率是2 cm-1.所有数据处理采用软件NGSLabSpec1-Origin中的Baseline correction进行处理,并用Origin 8.0工具作图.
3 结果与讨论
3.1 分子结构及优化
优化结果中没有发现虚频,表明优化得到的结构是5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的稳定构型(见图1).计算结果表明,优化后的分子结构对称性属于C2.金属钇原子被夹入卟啉环与酞菁环之间,形成三明治型结构,两个环扭曲成碟子状,一个碟子相对于另一个扭曲了45度[22],N6N8N9N7,N12N10N11N13的二面角均接近0.00˚,中心钇原子与酞菁环的四个异吲哚氮原子及卟啉环的四个吡咯氮原子配位形成四方反棱柱的配位结构(见图2),O代表四个异吲哚(吡咯环)氮原子平面的中心.由于引入了两个二茂铁基团,在卟啉环meso位发生取代,引起吡咯环的扭曲变形,与酞菁环之间空间位阻效应的相互影响,异吲哚也发生扭曲,伴随着两个二茂铁基团垂直于卟啉环各自相向旋转45度,卟啉环与酞菁环以金属钇原子为中心呈穹型围绕在其周围(见图3).
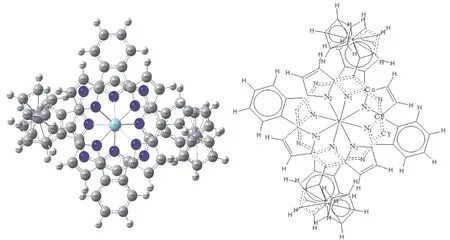
图1 5,15-二(二茂铁基)-卟啉酞菁钇分子结构模型示意图Fig.1 Optimized structure of[Por(Fc)2]Y(Pc)
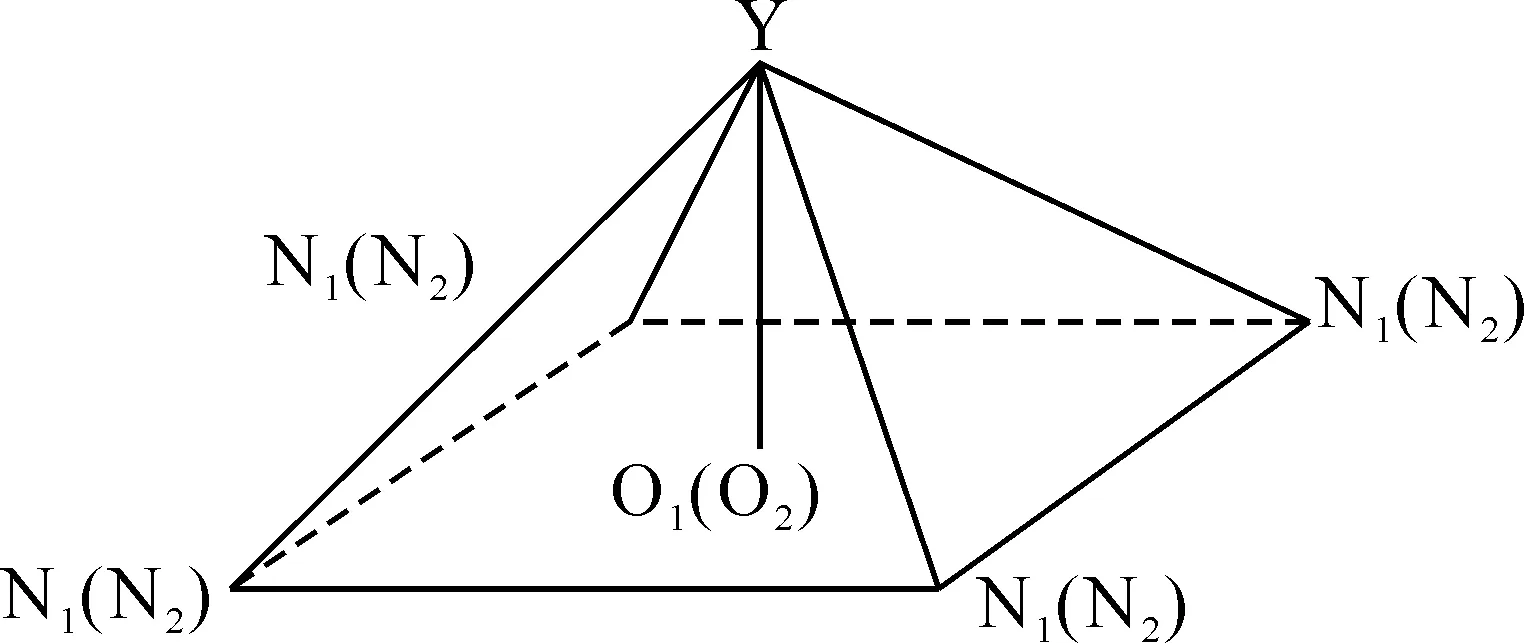
图2 Y原子与N原子组成的四方反棱柱体几何构型Fig.2 Geometrical configuration of tetragonal antiprism made of Y and N
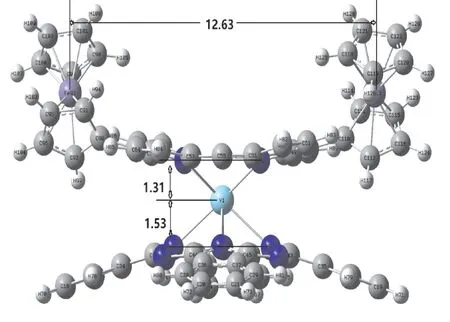
图3 5,15-二(二茂铁基)-卟啉酞菁钇部分原子间距离示意图Fig.3 Optimized distances between some atoms in[Por(Fc)2]Y(Pc)
3.2 5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的红外谱带振动归属
[Por(Fc)2]Y(Pc)分子由129个原子组成,共有381个简正振动模式.利用GaussView软件对5,15-二(二茂铁基)-卟啉酞菁钇的简正振动模式进行指认归属.本文选取[Por(Fc)2]Y(Pc)分子2000-400 cm-1的理论计算和实验数据作红外光谱图(见图4),理论计算和实验所得的红外光谱图基本上是一致的.对理论计算和实验光谱主要振动峰进行了线性回归拟合[23](见图5),结果表明,相关系数为0.992,标准偏差16.96,两者吻合较好,说明本文所选取的DFT理论计算方法以及水平是可行的.

表1 Y原子到N原子(异吲哚、吡咯环)平面的距离(Å)、主键长(Å)、键角(˚)Table 1 The calculated distances from Y to nitrogen plane(isoindole ring,pyrrole ring)and the main bond lengths and bond angles of[Por(Fc)2]Y(Pc).(Bond length unit:ÅBond angle unit:˚)
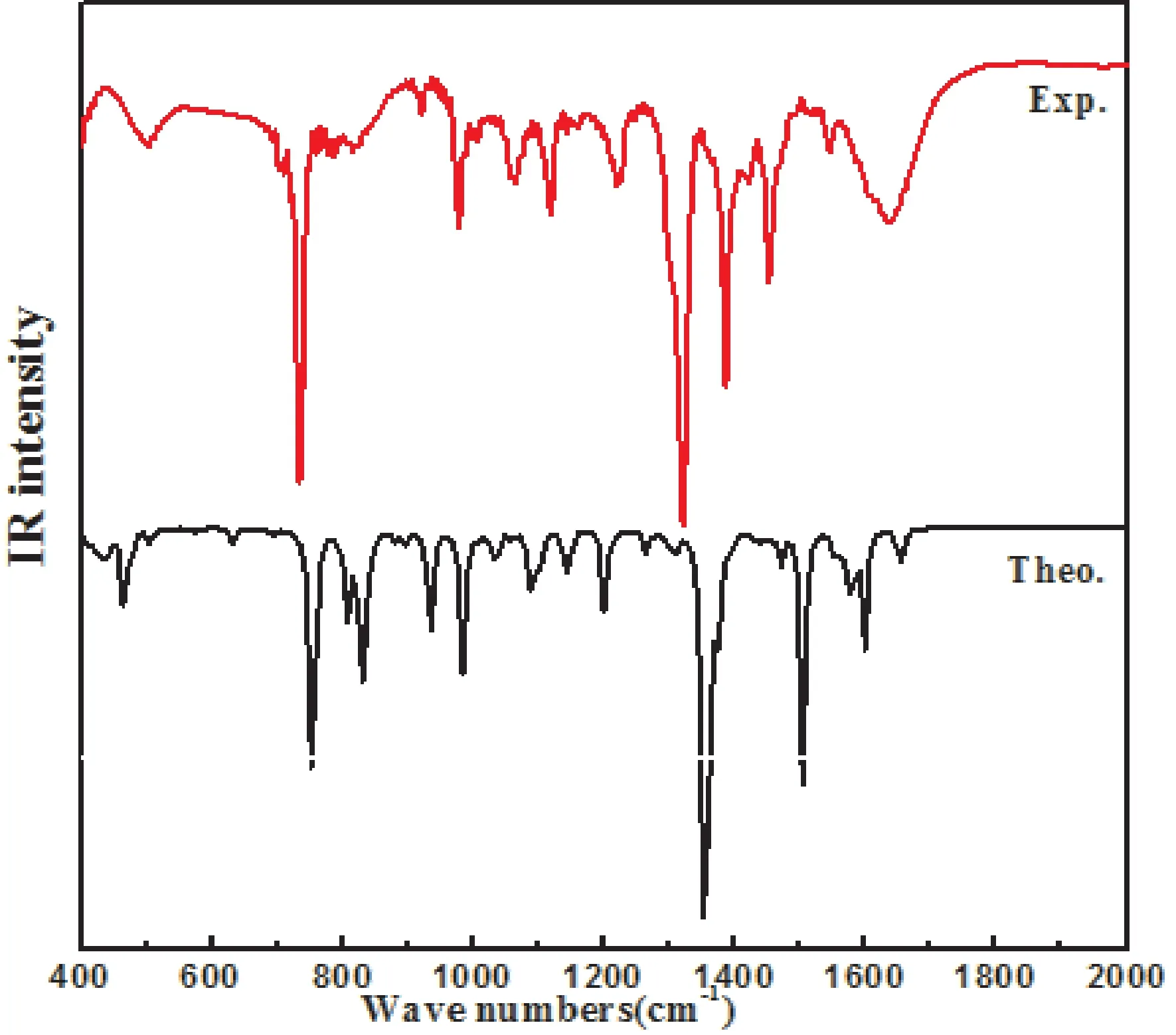
图4 [Por(Fc)2]Y(Pc)理论计算和实验的红外光谱图(红色为实验、黑色为计算)Fig.4 The theoretical(black)and experimental(red)IR spectrum of[Por(Fc)2]Y(Pc)
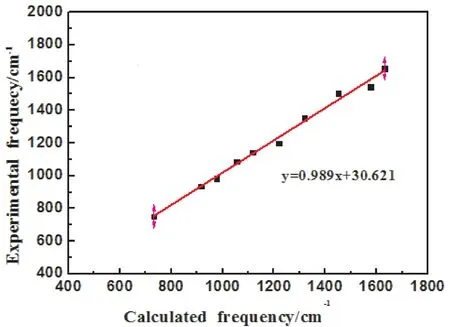
图5 [Por(Fc)2]Y(Pc)主要实验和理论计算频率线性回归拟合Fig.5 The linear regression fitting between main experimental and theoretical IR frequencies of[Por(Fc)2]Y(Pc)
通过对红外谱图分析466 cm-1的红外峰被指认归属于二茂铁茂环的面外摇摆以及吡咯环的面内摇摆振动;755 cm-1是红外谱图中的次强峰,被指认归属于异吲哚C-H面外弯曲振动,吡咯环C-N-C的面外弯曲振动;809 cm-1被指认为吡咯环C-H面外弯曲振动;830 cm-1归属于二茂铁茂环C-H面外弯曲以及吡咯环C-N-C的面内弯曲振动;935cm-1归属于二茂铁茂环C-H面内弯曲以及吡咯环C-H面外弯曲振动.985 cm-1被指认归属于吡咯环的C-H面内弯曲,C-N-C面内弯曲振动以及二茂铁茂环C-H面内弯曲振动;1201 cm-1被指认为吡咯环C-H面内弯曲,C-N-C面内弯曲振动;1355 cm-1是红外谱图的最强峰,归属于吡咯环C-C伸缩、CN-C面内弯曲振动以及异吲哚C-H面内弯曲振动;1374 cm-1的红外峰被指认为吡咯环的C-N-C面内弯曲振动,异吲哚C-H面内弯曲振动;1505 cm-1是红外谱图的较强峰,被指认归属于异吲哚C-H面内弯曲振动;1599 cm-1红外峰被指认为吡咯环C-C伸缩,C-H面内弯曲振动.
3.3 5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的分子表面静电势
空间中某一点的静电势是指将单位正电荷从无穷远移动到该点所需的功.在原子核和电子共存的分子系统中,原子核和电子都会影响分子的静电势.因此,分子周围空间与原子核距离不同的点静电势也不同.静电势对于研究分子之间的静电相互作用,预测反应位点和预测分子性质具有重要意义.分子静电势图上的不同颜色表示具有不同静电势的区域.其中,红色,黄色,绿色和蓝色表示电负性逐渐降低.蓝色区域具有更大的负静电势,而红色区域具有更大的正静电势.白色区域的静电势接近零.红色区域表示带负电的区域,即电子的密集区域,该区域更容易受到亲电试剂的攻击.蓝色区域表示带正电荷的区域,更容易受到亲核试剂的攻击.分子表面静电势分布图[24](见图6)分析结果表明,5,15-二(二茂铁基)-卟啉酞菁钇分子存在极小值点34个,最小点位于吡咯环上的N13原子处;极大值点53个,最大点位于异吲哚苯环上与C19相连的H71原子上.
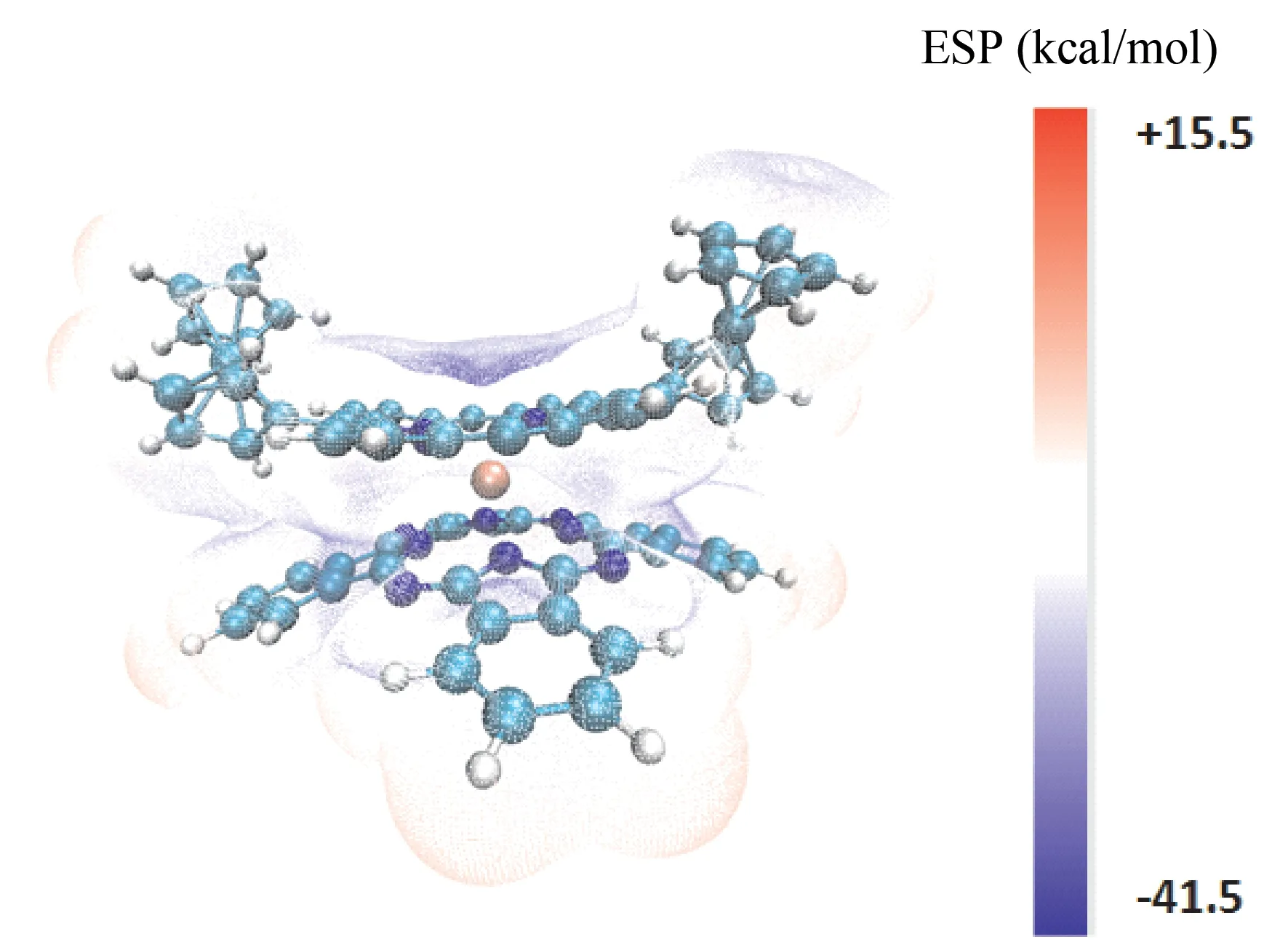
图6 (Pc)Y[Por(Fc)2]分子静电势图Fig.6 Molecular electrostatic potential diagram of[Por(Fc)2]Y(Pc)
4 结 论
选用混杂的B3LYP密度泛函方法,在Lanl2dz水平下,对5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的结构进行了优化,对分子内主要的键长与键角进行了计算,通过频率计算,获得了5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的红外光谱,理论计算与实验所得的红外光谱图基本上是一致的,对理论计算和实验光谱主要振动峰进行了线性回归拟合,相关系数为0.992,标准偏差为16.69,两者吻合较好,说明本文选取的DFT理论计算方法是可靠的,并结合GaussView软件对5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的简正振动模式进行了指认归属.此外,分析并讨论了5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)的分子静电势.对研究5,15-二(二茂铁基)-卟啉酞菁钇[Por(Fc)2]Y(Pc)分子的性质,提供了理论基础.
