An overview of the complexity of DNA storage
2021-08-16Linhuin
-, -, Lin, , -hu, -in*
(a. Institution of Computational Science and Technology; b. Institution of Huangpu Research, Guangzhou University, Guangzhou 510006, China)
Abstract: DNA has become a potential new storage medium with its high data density, long storage time and low maintenance cost. At present, the development of DNA storage technology is still facing several challenges: ① far higher than the error rate of traditional storage technology; ② The distribution of DNA storage molecules was not uniform; ③ Loss of storage molecules DNA has become a potential new storage medium with its high data density, long storage time and low maintenance cost. At present, the development of DNA storage technology is still facing several challenges: ① far higher than the error rate of traditional storage technology; ② the distribution of DNA storage molecules was not uniform; ③ loss of storage molecules. In this review, for better understanding of the complexity of DNA storage, we first provide the framework of DNA storage including DNA synthesis, PCR, and sequencing. Then we analyze the errors and molecular bias, as well as the sequence loss during DNA storage. Finally, we make a conclusion about the complexity of DNA storage and envision a more robust DNA data storage system which requires both focus and research efforts from various fields.
Key words: DNA storage; errors; molecule bias; sequence loss
As a burgeoning data storage media, DNA have inherent advantages compared with traditional storage media like hard disc, compact disc and magnetic tape. First, the longevity, in 2015, Grass, et al.[1]demonstrated that data carried in DNA molecules can be preserved for thousands of years, while current available storage technologies guarantee data integrity for only several years. Second, the enormous information density, which is theoretically up to 2 bit per base, means that all the data throughout human history can be stored in a garage sized space. Third, low maintenance costs, the environmental configuration for storing data carried in DNA molecules is easy to implement with the current technological progress. These three features make DNA storage a promising research field[2].
Besides, we also have to endure the defects of DNA storage. Although DNA synthesis technology has experienced the development from deoxy-polynucleotide synthesis[3]to synthesis chip technology[4]and de novo DNA synthesis using polymerase-nucleotide conjugates[5], indicating synthesis technology is becoming a higher-throughout technology, it is still very hard to synthesize a DNA sequence with a length of over 300 bases dues to the constraint of synthesis technology, and data can only be written into many short sequences and stored in a pool in an unordered way, therefore data cannot be randomly accessed. Besides, the development of synthesis technology introduces more errors into DNA sequences, it is an error-prone technology. Furthermore, accessing the data requires sequencing technologies which also experience the development of three generation including Sanger[6], Illumina[7]and nanopore[8]. Similar to synthesis technology, the nanopore has the highest-throughput and highest error rate compared with the old sequencing technologies. As sequencing(reading) process amounts to random sampling from the sequence pool and reading the samples, it cannot be ensured whether the samples we take contain all the sequences required by the data recovery or not, if not, we call this problem a sequence loss. Usually, the sequencing process is preceded by several cycles of PCR. However, all of the three main processes of DNA storage including synthesis, PCR and sequencing are error-prone and result in a very uneven distribution of DNA molecules. In practice, the length of data carried in a DNA molecule is 160~180 bases, and the error rate is 1%~2%[9-11], that means nearly every data sequence carried in DNA has at least one error. Moreover, approximately 88% of sequences in a DNA sequencing file have an incorrect length due to three types of error such as insertion, deletion or substitution[12]. Meanwhile, to offset errors within sequence, molecule bias and sequence loss, logical and physical redundancy is introduced into a DNA storage system, resulting in an extremely large sequencing file. For example, a 17 Mb sized original file corresponds to 1.3 Gb sized sequencing file. The above problems reflects the complexity of DNA storage. We summarize the complexity in DNA storage to ‘Errors’, ‘Bias’ and ‘Sequence loss’ and will discuss in detail in section 2, 3 and 4.
As a cutting-edge interdisciplinary technology, DNA data storage has attracted much focus and many research efforts from various fields. In 2012, Church, et al.[13]tried to store about a megabyte of data in DNA without a principled way of error correction, therefore failed to decode it back into original digit data. In 2015, Yazdi, et al.[14]achieved selectively accessing files. In 2018, Organick, et al.[12]stored more than 200 megabytes of data in DNA and successfully retrieved which is the largest scale of data stored in DNA. However, the costs, the accessibility of synthesis and sequencing technology still have to advance significantly for DNA data storage to become a more common data storage technology. Unfortunately, the advancement in technology will most probably lead to lower precision. In the future, the error correcting scheme in DNA storage will become more important since perfect data recovery will become harder.
1 The framework of DNA storage
Studying the complexity of DNA storage requires a comprehensive understanding of each process during DNA data storage, since all the molecule bias and errors including insertion, deletion or substitution are introduced by these processes. DNA data storage technology stores digital data via synthetic DNA[15]. Data writing corresponds to encoding the digit data into nucleotide sequences and synthesizing the corresponding single stranded DNA molecules. The synthetic DNA molecules are kept in an appropriate environment[16-17]. Data reading corresponds to sequencing the data carried in DNA molecules and decoding it back into the original digital data. Moreover, some necessary DNA processing steps are also required during DNA storage.
For the purpose of understanding the complexity of DNA storage, we provide the framework of DNA storage including three main steps which are synthesis, PCR and sequencing before discussing the complexity of DNA storage, just as illustrated in Fig.1. We also make a comparison for the three generations of DNA synthesis and sequencing technology in Table 1. The detail of the framework is as below.
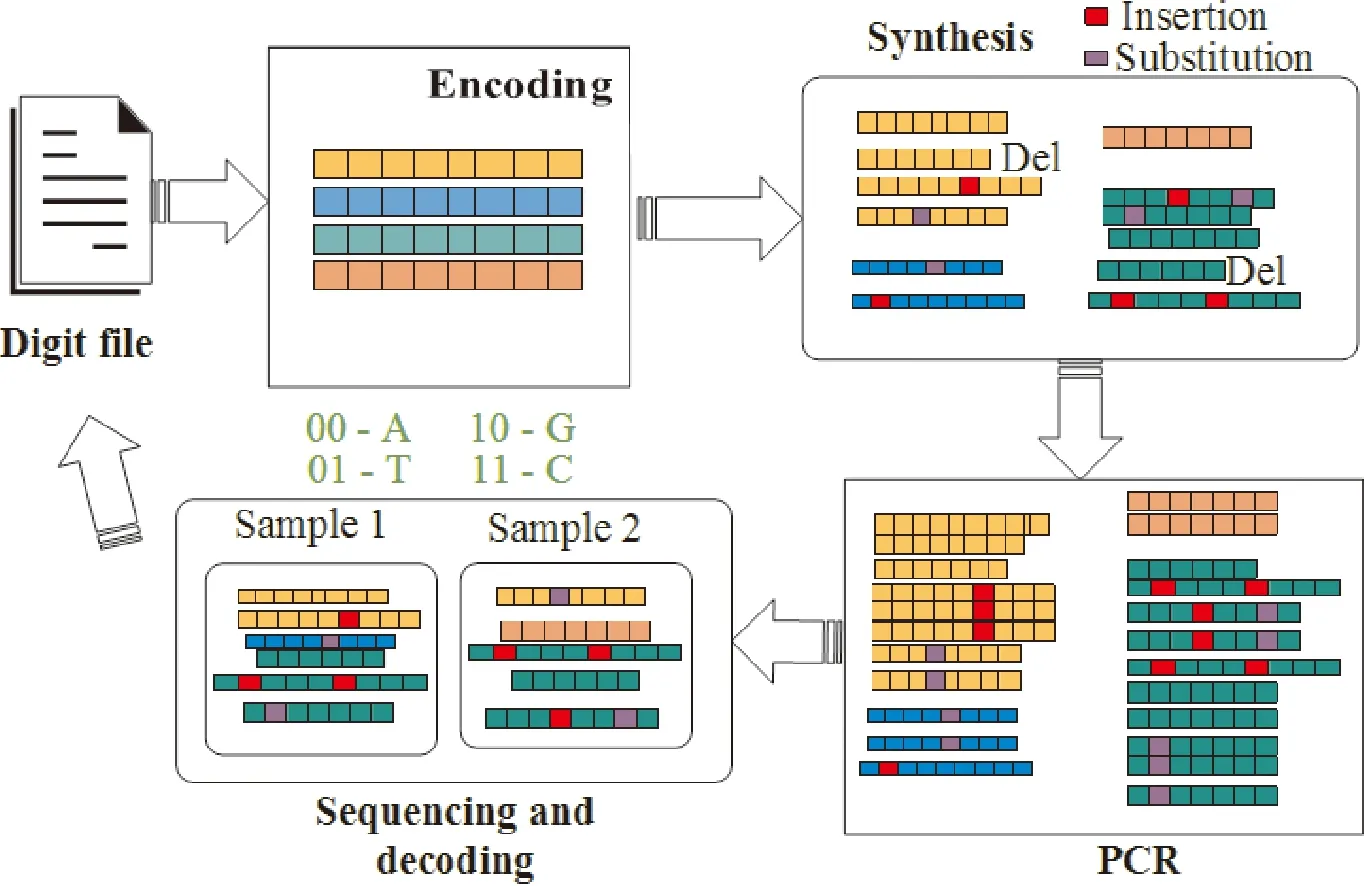
Fig.1 The framework of DNA storage
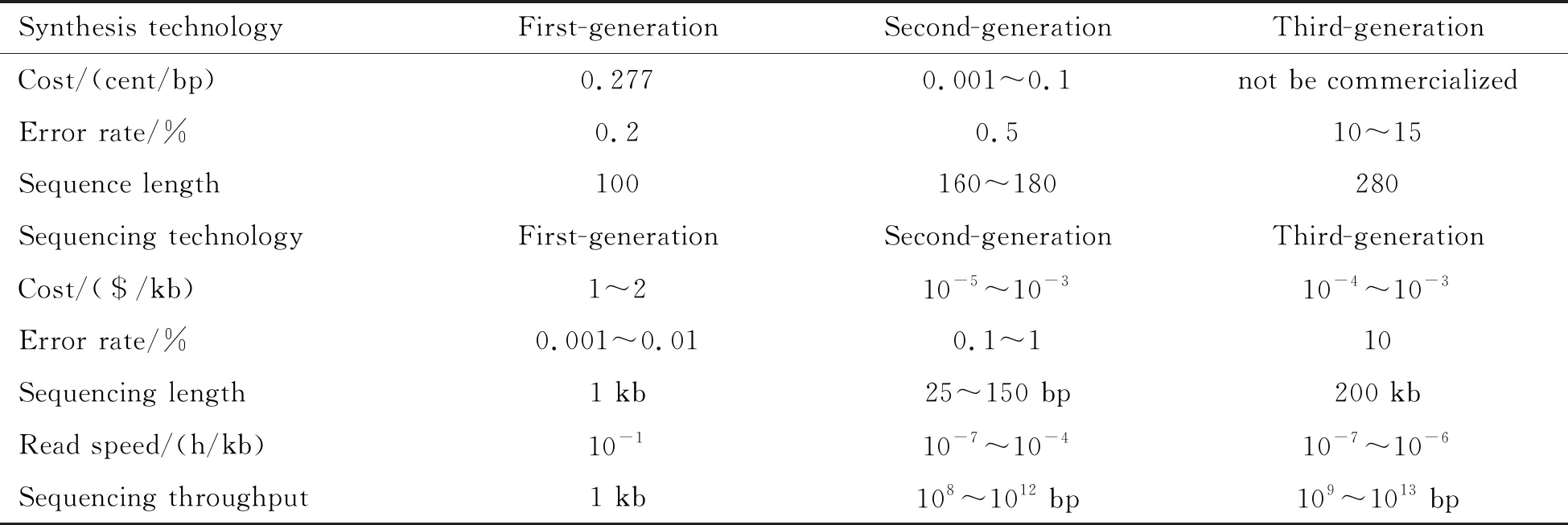
Table 1 A comparison between the three generation of DNA synthesis and sequencing technologies
1.1 DNA synthesis
Most of the recent works of DNA data storage use synthesis chips to synthesize data carried in DNA molecules. It is a high-throughput technology but has more errors than deoxy-polynucleotide synthesis. Since the de novo DNA synthesis using polymerase-nucleotide conjugates has not been commercialized, DNA chip synthesis technology is the best choice now.
This step amounts to writing the data into traditional storage media. Since a DNA molecule consists of four bases adenine(A), thymine(T), guanine(G), cytosine(C), the digit data have to be encoded into nucleotide sequences first. Due to the technology constraints of synthesis, a nucleotide sequence with a length over 200 bases is very hard to obtain in practice. Usually, in DNA storage a digital file corresponds to many short nucleotide sequences with a length range 160~180.
Current synthesis technology works on solid surfaces called chips, on the one end of the chip the DNA molecules are attached. Through a specific chemical reaction, the nucleotides are added onto DNA molecules[18], resulting in growing sequences. Current synthesis technology only generates single stranded DNA molecules. Besides, for a specific sequence, the synthesizer generates millions of sequence copies[19].
Synthesis technology is not perfect. First, during the chemical reaction of adding nucleotides onto a growing DNA molecule, three types of errors (insertion, deletion or substitution) might occur. Second, the chemical reaction might terminate[20], as a result, some DNA molecules do not reach the target length, these sequences are called non-complete sequences. Third, the distribution of synthetic DNA molecules is very uneven, this is due to the different synthesis yields on synthesis chips[21], some location on chips intrinsically have higher yields than others, also some locations intrinsically have lower yields than others.
1.2 Polymerase chain reaction (PCR)
Polymerase chain reaction (PCR) is implemented to amplify the DNA molecules and prepare for DNA sequencing. It is a crucial step in DNA data storage having a great relevance to molecule bias.
As described above, DNA synthesis is error-prone, generates a single stranded DNA sequence and non-completed sequence. In practice, given a digital file, the corresponding DNA sequences must contain logical redundancy (primers, error correcting codes and indices) in both ends to enable data recovery, and non-completed sequences probably contain no primers at both ends. According to this, PCR is first implemented in DNA storage to generate double stranded DNA molecules and clean up the synthesis pool, for DNA molecules containing no primers at both ends can not be amplificated by PCR and thus be diluted.
Since the utility of PCR depends on primers, in 2018, a group led by Organick, et al[12]devised a primer library containing thousands of pairs of orthogonal primers enabling specific file recovery, so that they can be randomly accessed in DNA storage. Their design criteria of primer sequence is as follows: The maximum of consecutive As(Ts) and Gs(Cs) is 3 and 2 respectively, the maximum of self-complementarity and inter-sequence complementarity is 4 and 10 respectively, GC content is 45%~55%, the minimum Hamming distance between each other is 6.
Moreover, PCR must be implemented many times, since sequencing amounts to sampling from sequence pool and reading the samples, the uneven distribution of DNA molecules in the synthesis pool may lead to sequence loss and impact decoding back into the original file, so before sequencing we implement PCR to amplifiy the DNA molecules in the synthesis pool to increase the physical redundancy[22], therefore, constructing a more robust DNA storage system.
Same as DNA synthesis PCR is also a paramount source of molecule bias[21], in each cycle of PCR, each sequence has a specific amplification factor range 1.6~1.8, that means if we implement large cycles of PCR, the distribution of DNA molecules in the original pool may become more uneven, eg:1/1-(1.6/1.8)10=0.004 5. We should also note the original synthesis pool is not an even distribution. Along with DNA synthesis, implementation of PCR in DNA storage will further aggravate the uneven distribution of DNA molecules. Significantly, PCR by itself is known to be a high-fidelity process, therefore during the implementation of PCR, seldom errors will occur[23]. Details about molecule bias and errors will discuss in section 2 and section 3.
1.3 Sequencing
The current two mainstream sequencing platforms are Illumina and nanopore. Since nanopore is a newly emerging sequencing technology, most of the DNA storage readout is performed by Illumina. Given a DNA sequence, we can get many short reads of the sequence via sequencing technology, then we assemble these reads together and decode it back into the original digital file.
Sequencing amounts to sampling from the DNA pool and reading the sequences in the samples, so only a small fraction of the DNA molecules in the pool can be obtained. Note that after synthesis and cycles of PCR, the molecule distribution is very uneven and errors might occur within every sequence, the quality of the obtained fraction depends on molecule distribution in the pool and the number of samples we take. Loss of sequences and errors within sequences in the obtained fraction may lead to failure in data recovery.
Same as synthesis, sequencing technology is also error-prone, this process itself may also introduce errors within the DNA sequence. Moreover, sequencing errors are not random, according to Ref.[24], it is strand specific, and substitutions predominate[25].
The framework of DNA storage is very important for understanding the complexity of DNA storage, since the complexity refers to molecule bias and errors within the DNA sequence, and both of them are mainly related to the three main steps in this framework. Meanwhile, some necessary processing of DNA during DNA storage will also have impact on the complexity in DNA storage, the next two sections will discuss molecule bias and errors within the DNA sequence in detail.
2 Errors
DNA storage is an error-prone storage system, the reading and writing error rate of traditional commercial hard drives are as low as 10-15, while the error rate of DNA synthesis is generally 1/200 to 1/2 000, and the error rate of next-generation sequencing is 1/100 to 1/1 000[26], indicating inefficiency and less reliability in DNA storage.
There are three types of errors: insertion, deletion and substitution. Insertion refers to a nucleotide being placed where it should not be, deletion refers to a nucleotide being absent, substitution refers to adding an unexpected nucleotide rather than the intended one. As discussed in section 1, the insertion, deletion, and substitution errors are mainly introduced by synthesis and sequencing.
2.1 Sequencing and synthesis error
From section 1 we know synthesis and sequencing are paramount sources of errors within DNA molecules. In practice, we can only estimate the sequencing error independently, due to its two-sided reads. The Ref.[27] estimated the sequencing error probabilities by aligning the single sided reads (obtained from two-sided reads) which are successfully aligned by FLASH[28]and find the substitution errors predominate the sequencing error. Moreover, in overall error probabilities the deletion and insertion predominate. According to this, they infer that deletions and insertion are mainly due to synthesis while substitutions are mainly due to sequencing.
2.2 Errors during storage
After synthesis and before sequencing, DNA molecules are stored in a DNA pool. Some necessary processing such as removing molecules from synthesis chips and, heating intervals in PCR, may lead to DNA decay, and the effects of decay include strands breaking, cytosine deamination which resulting in U-G base-pair, and will further resulting in translation of C to T and G to A. Moreover, DNA aging[29]significantly increases the substitution errors while having little influence on insertions and deletions.
According to the above description we can conclude the deletions and insertions predominate in overall error probabilities, and are mainly due to DNA synthesis, while substitutions predominate in sequencing and DNA decay during storage also contributes to substitution errors.
2.3 Effects and solution of errors within DNA molecules
In practice, data carried in DNA molecules typically has a length of 160~180, and error rate is 1%~2%. Statistically, nearly every sequence has at least an error, and most of them probably have 2~3 errors. Fig.2 shows the distribution of errors within a molecule. It is reported 88% of sequences in the sequencing file have incorrect length[12]. All of these make DNA storage an unreliable and inefficient system. To recover the original file from a DNA sequences, we have to handle a sequencing file which contains many irregular sequences, we need to detect the errors within sequences, and then try to recover it. In practice, the work of data recovery is known to be a complicated task.
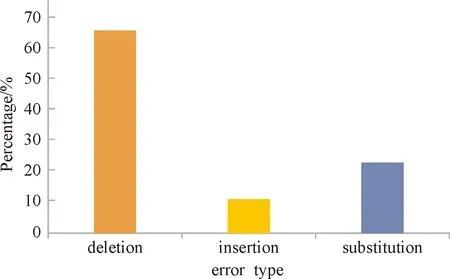
Fig.2 The proportion of different errors in sequencing file
We summarize the solution of errors to repetition and error correcting code. Repetition is easy to implement, such as increasing the number of PCR cycles, raising sequencing coverage, then there are enough repetitions of every sequence to ensure data recovery. However, simply relying on repetitions of sequences to solve the problem is not the optimal scheme, because the cost is high and the sequencing file may become redundant, as a result, the storage system turns out to be inefficient and complicated, moreover, previous studies proved that the intrinsic data redundancy cannot guarantee perfect data recovery[16,29].
So many error correction schemes have been proposed, and all of these codec schemes require logical redundancy within the DNA sequence. In practice, a reliable codec system contains two layers: inner code and outer code. The inner code is used to correct the errors within the DNA molecule while the outer code is implemented to handle the sequence dropouts. For those sequences that cannot be corrected by correcting code will be deleted. A group led by Grass introduced the Reed-Solomon erasure code which can solve the substitution errors[30]. However, for deletion and insertion errors, which is quite different with traditional storage medium and difficult to deal with[31], Press et al. proposed a coding scheme based on hash code a greedy exhaustive decoding scheme[32]. The scheme can correct insertion and deletion errors within single stranded DNA molecules, but it needs high redundancy to achieve error correction, and the complexity of decoding is very high. Sabary, et al.[33]proposed a dynamic DNA reconstruction algorithm, which can reconstruct DNA sequence under high error rate. Song, et al.[34]designed a highly robust DNA sequence reconstruction algorithm based on e-bruijn graphs, which can quickly reconstruct DNA fragments from multiple error-rich sequences such as insertion, deletion or substitution errors. Many researchers also study in vivo DNA storage[35-41], which is high fidelity and guarantees long term data replication. Usually, correcting 2~3 errors requires 5~10 logical redundancies which will increase the synthesis cost nearly 10%, compared with repetition scheme, error correct code is economical and practical.
We hope DNA storage becomes a more common data storage technology in the future, to achieve this goal, the costs and accessibility of DNA synthesis and sequencing must be advanced significantly which will probably result in lower precision and introduce more errors to the DNA sequence. The need of an effective error correcting scheme is very urgent.
3 Bias
According to the description of DNA storage framework, the two paramount sources of molecule bias in DNA storage are synthesis and PCR. This section will first focus on the two main sources of molecule bias in DNA storage as well as the bias introduced by some necessary processes during DNA storage, then we will discuss the resource waste and inefficiency it may bring to the DNA storage system.
3.1 Synthesis bias
A recent study revealed the synthesis bias is related to the spatial location on the synthesis chip[21]. In their experiment, the synthesis pool contained 1 536 168 unique DNA sequences synthesized via Twist Bioscience. And all sequences in the pool already contain adapters and primers required by Illumina sequencing, so that sequencing can be implemented without sequencing preparation like PCR. Then they map the sequencing reads back to the responding position on synthesis chip and observe a distinct pattern, thus demonstrating their inference.
Besides, DNA synthesis is a chemical reaction, the specific chemical reaction enables nucleotides added one by one onto the growing sequence which then attach to the synthesis chip. However, the termination of chemical reaction may happen during DNA synthesis, resulting in many non-completed sequences. Due to a lack of primers at the both ends, the non-completed sequences will be diluted, and the proportion of corresponding sequence will decline.
3.2 PCR bias
Many previous studies have observed molecule bias during PCR[42-44], there are three main sources of PCR bias: GC content, primers on both ends of a sequence and PCR stochasticity.
In 2012, Ref.[43] reports the obtained coverage of sequences with GC content smaller than 20% or larger than 75% is significantly lower than others, this finding indicates PCR prefers sequences with GC content between 20%~75%. PCR bias is related to primers due to the presence of non-completed sequence after DNA synthesis. PCR stochasticity is that in each cycle of PCR, each sequence has a specific amplification factor range 1.6~1.8, if the number of cycles is large enough, eg:1/1-(1.6/1.8)10=0.004 5, we will observe a very uneven distribution of DNA molecules in a DNA pool.
Since primer in a specific DNA sequence depends on synthesis, we have to figure out the paramount source of PCR bias in GC content and PCR stochasticity. In Ref.[21] they designed an experiment to study the GC bias. They used two groups of data, the first group was encoded to refrain from homopolymers, the GC content in this group was between 40% and 60%, the number of PCR cycles was 31, then they observed a significant change between population fraction and GC content in statistic, but it did not mean there was a close link between population fraction and GC content, because the slope of the linear fit is very small (<0.01). The second group was encoded allowing random homopolymers, the GC content ranges 25%~75%, still the small slope of linear fit (<0.01) indicates few associations between population fraction change and GC content. According to this they concluded that PCR stochasticity predominates the PCR bias.
The impact of GC content in PCR bias attributes to the effect of hydrolytic damage during DNA storage, the main effects include depurination[45]and deamination of cytosine which result in strands breaking[46]and U-G base pairing[47]respectively. Once a sequence undergoes strand breaking it can no longer be sequenced by sequencing technology, as described in section 1 sequencing technology involves PCR and broken strands containing no primers at both ends cannot be amplified by PCR, therefore they cannot be sequenced. Moreover, different enzymes have different effects on U-G base pairs during PCR. Strands containing U cannot be amplified if the Proof-reading enzymes are used by PCR, and any completed sequence can be amplified by using non-proof-reading enzymes, but in subsequent PCR, C will be transformed into T and G will be transformed into A. Note that the above effects are less extreme in PCR bias.
3.3 Other sources of bias
Except two paramount sources of PCR bias, some necessary processing and chemical reactions also introduce bias to DNA storage. Such as removing DNA strands from synthesis chips and heating intervals in PCR, will lead to DNA decay. The most frequent form of DNA decay is hydrolytic damage, the effect of hydrolytic damage is strands breaking or cytosine deamination (G-U base pair). The breaking DNA strands and strands containing G-U base pairs cannot be amplified by PCR, thus being diluted in subsequent processes.
3.4 Effects and solutions of molecules bias to DNA storage
Molecule bias in DNA storage may lead to serious resource waste. To recover the original data, usually we sample from the DNA pool, due to the uneven distribution of DNA molecules, some sequences in samples we take may have significantly more copies than others while some may have fewer copies than others, Fig.3 shows this problem. In practice, the first situation may cause serious resource waste, because the sequencing platform has to handle the same sequence many times and further results in a very gluttonous sequencing file, then we also have to handle the gluttonous sequencing file to recover the original file, which is time and space consuming. The second situation makes the data recovery become difficult, fewer sequence copies means less decoding capacity, since the error may occur in every sequence and some sequences may contain significantly more errors, more numbers of copies ensure larger decoding capacity. Choi, et al.[48]implemented in situ amplification, after 20 cycles of amplification, no obvious change in the distribution of DNA fragments was found, which significantly reduced the problem of molecule bias. Gao, et al.[49]realized low bias and stable amplification by preference and stable repetition by isothermal chain replacement amplification.
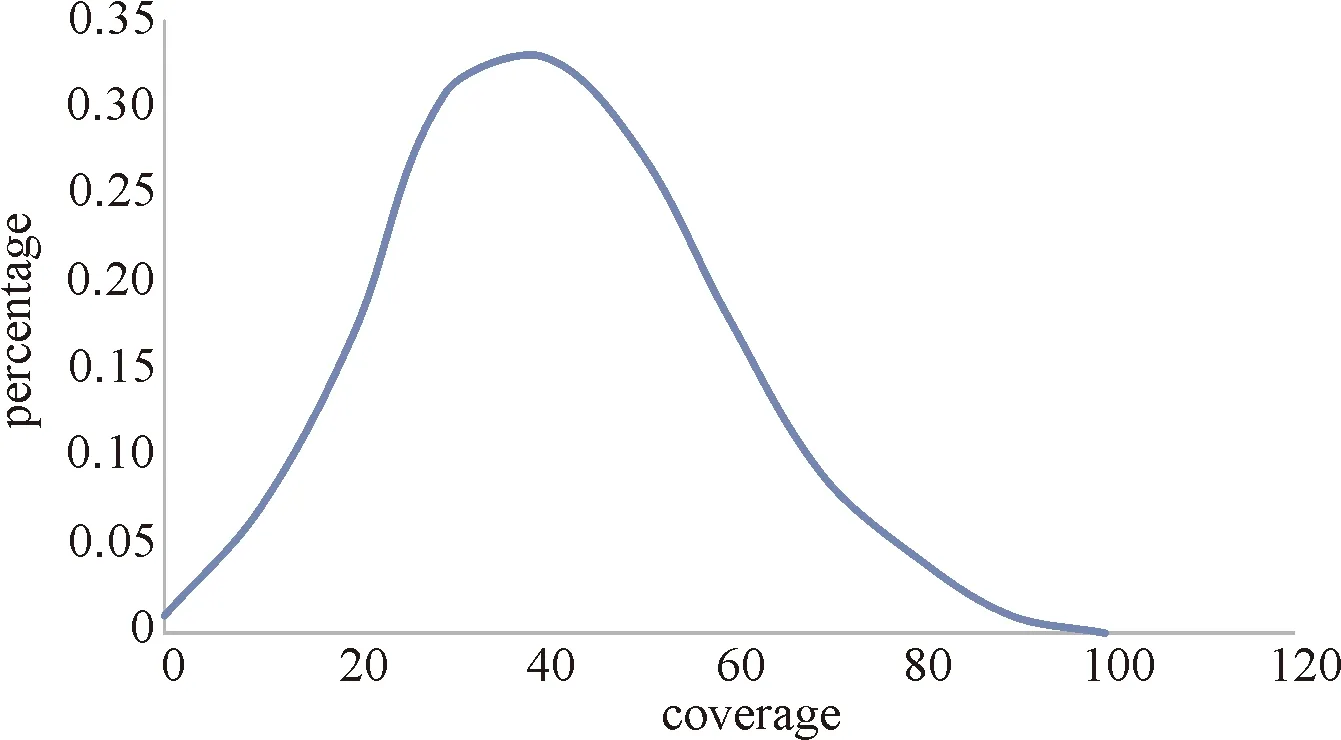
Fig.3 The sequence distribution of sequencing file
However, we still expect a more robust DNA storage system with less molecule bias, it requires focus and research efforts from various fields to make great progress in DNA synthesis, sequencing and PCR technology.
4 Sequence loss
Unlike traditional storage medium, the data carried in DNA molecules are stored in a DNA pool without spatial order, as a result, the way of accessing data is quite different. In DNA storage, we sample from the DNA pool, and the quality of the samples we take depends on DNA molecule distribution in the DNA pool and the number of samples we take. However, no matter how even the molecule distribution and how large the number of samples is, it still cannot be ensured that all the sequences required by data recovery are included in the samples we take. In practice, a common situation is some sequences may be lost during sequencing. Although we can take more samples from the DNA pool once sequence loss happens, this way is far from optimal. Simply taking more samples will significantly enlarge the sequencing file since in DNA storage a 17 Mb sized digital file corresponds to a 1.3 Gb sized sequencing file, and decoding the DNA sequences back into the original digital file is through the sequencing file, handling a large sequencing file is time and space consuming.
Similar with the errors within DNA sequence, the Digital Fountain code[50]and Reed-Solomon code are designed to overcome the problem of sequence loss. Reed-Solomon code tolerates a finite numbers of sequence loss, but the cost of data recovery and data update is high. Digital Fountain code has a simple but nonlinear encoding/decoding scheme and has lower decoding cost compared with Reed-Solomon code. However, a more advanced codec systems which requires low cost and linear is still unavailable.
5 Conclusion
In this review, we characterize the complexity of DNA storage as errors within the DNA molecule, molecule bias and sequence loss. Through the providing framework of DNA storage as well as the thorough analysis about every possible problem during DNA storage, we can make conclusions as follow.
We conclude that for errors within DNA molecules, synthesis and sequencing are two paramount sources, the decay and aging of DNA mainly introduces substitutions to a DNA molecule. In overall error probabilities, the deletions and insertions predominate, while substitutions predominate in sequencing error. And molecule bias in DNA storage is mainly due to DNA synthesis, PCR, the decay of DNA during storage also has an impact on bias. Moreover, the synthesis bias is due to the different synthesis yields on a synthesis chip, and PCR bias is mainly due to PCR stochasticity, for each cycle of PCR every sequence has a specific amplification factor range 1.6~1.8, also PCR has a preference of GC content range 25%~75% and will dilute the non-completed sequence produced by synthesis. Meanwhile, the decay of a DNA molecule may result in strands breaking and be diluted in subsequent steps. Finally, sequence loss in DNA storage can be solved by Reed-Solomon code and Digit Fountain code, but an economical and efficient codes scheme is still needed.
All in all, impelling DNA storage to become a common storage technology, both hardware and software need great improvements. Although the development of synthesis and sequencing technologies tends to be lower cost, higher speed and throughput, it also becomes more error prone. Many codec schemes have been proposed to resist errors, bias and sequence loss, but DNA storage is still a high cost, inefficient storage medium compared with traditional storage styles. I wish more improvements about reducing error rate and bias on synthesis and sequencing technologies, also the decoding capacity and decoding/encoding costs have a great potential to develop. Finally, We hope this review can attract more focus and research efforts from various fields to build a more robust DNA storage system in the future.
