CaO基吸附剂捕集CO2及其抗烧结改性研究进展
2021-08-15耿一琪郭彦霞程芳琴成怀刚
耿一琪,郭彦霞,樊 飙,程芳琴,成怀刚
(山西大学 资源与环境工程研究所 CO2 减排与资源化利用教育部工程研究中心,山西 太原 030006)
CO2是一种常见的碳氧化合物,大气中的CO2可以吸收地表放出的长波热辐射,使地表附近气温升高,产生温室效应,CO2对温室效应的贡献率可达70%[1]。温室效应导致的全球变暖正在带来一系列的气候灾害,如南北两极冰川融化、极端天气发生、海平面上升和海洋酸化等。当大气中CO2体积分数达到约1.0×10−3时,海洋表面温度上升将导致海洋的碳吸收量减少9%−15%,对大气中CO2浓度的上升产生正反馈作用[2],如果不加以控制,CO2过高带来的气候问题将愈演愈烈,形成恶性循环。截止到2019 年,美国国家海洋和大气管理局通过莫纳罗亚气象台监测到大气中CO2体积分数已经超过4.15×10−4[3],是百万年来地球CO2的最高。Andrew 等[4]采用IPCC 的IS92a 排放场景,预测到2100 年CO2体积分数将达到7.9×10−4,北半球和热带地区温度将升高4.5℃。
CO2的人为排放主要产生于燃煤燃气发电、钢铁及水泥制造等工业生产过程,工业生产过程对大气中CO2增量的贡献率可达80%[5],控制工业CO2的排放是缓解全球气候变暖的关键。碳捕集、利用与封存(CCUS)技术是指将CO2从主要排放源中分离捕集出来,然后运输到指定地点,进行地质贮存或回收利用,被认为是实现二氧化碳减排的重要战略举措。CO2捕集是CCUS 技术最重要的步骤之一,即从大型污染源捕集CO2,这对CO2的减排至关重要,是整个CCUS 技术中成本最高、技术突破最困难的一步。CO2的捕集可以分为燃烧前捕集、富氧燃烧和燃烧后捕集三种。燃烧前捕集是对燃料进行预处理,使其在气化炉中形成以CO、H2为主的合成气,然后将合成气中的CO 转化为CO2,并对其进行捕集,得到富含H2的燃料气。燃烧前捕集的典型方法是整体煤气化联合循环技术(IGCC)[6]。燃烧中捕集也叫富氧燃烧,即通过空气分离装置分离出氮气,用纯氧气代替空气进行燃烧,可得到高含量的CO2,经捕集后可直接利用或贮存[7,8]。燃烧后捕集是指从燃烧后的烟气中捕集CO2,多适用于燃煤或燃气发电厂。燃烧后捕集是最成熟的CO2捕集技术,采用燃烧后捕集方法的优点在于不需要对电厂等大型排放源进行大规模改造,在其原有的基础上进行改建即可。大多数燃煤发电厂出口烟气温度在873 K 以上[9],高温固体吸附剂可以从高温烟气中直接捕集CO2而不需要降温处理,对降低能耗有重要意义,因此,高温吸附剂在CO2捕集中的研究和应用得到了广泛的关注。
CaO 基吸附剂是一种高温吸附剂,适合从高温烟气中捕集CO2,生成的产物CaCO3的分解温度约为825℃以上,经过CO2热脱附后,可以重新用于吸附CO2。同时,CaO 是碱性金属氧化物,对酸性气体CO2的吸附能力强(理论上,1 g CaO 可以捕集0.786 g CO2)。从吸附剂的来源看,煅烧来源广泛、价格低廉的石灰石等物质制备CaO 基吸附剂时会产生气体,得到高孔隙率的CaO 基多孔吸附材料,无需进行特殊的造孔改性处理。从捕集成本来看,CaO 基高温吸附剂虽在吸脱附过程能耗较高,但可以通过热量回收来提高其能量效益[10]。与典型的常温MEA 溶剂吸收法相比,其分离烟道气中的CO2需经过一系列的降温处理,会给火电厂带来相当于净发电量13%−37%的能量损失,且该方法回收和贮存CO2的费用高达50−60 美元/吨[9]。经综合计算,CaO 基吸附剂循环捕集CO2的成本为15−30 美元/吨,明显低于胺溶液吸收法(39−96 美元/吨)[11]。CaO 基吸附剂捕集CO2是一种有良好应用前景的技术。
CaO 基吸附剂可通过碳酸化/煅烧的吸脱附循环实现循环捕集CO2,在IGCC 的煤气化过程[12]、直接燃烧发电后[13]以及甲烷蒸汽重整制氢[14]等过程的CO2捕集方面均有广泛的适用性。然而,在循环捕集过程中吸附剂的吸附能力会因磨损、烧结、CaCO3产品层的覆盖等因素逐渐下降。其中,烧结是重要的影响因素之一,使CaO 基吸附剂的比表面积减小、捕集能力显著下降。近年来,越来越多的研究者开始关注CaO 基吸附剂的烧结行为、烧结机理以及抗烧结改性等方面的研究,以提高其吸附容量及循环稳定性。本文对CaO 基吸附剂的吸附原理、烧结失活机理及抗烧结改性方法进行了全面的综述,有望为高性能CaO 基吸附剂的研发提供重要的理论和技术支撑,同时为广大研究者进一步认识CaO 基吸附剂的技术进展及发展方向提供参考。
1 CaO 基吸附剂吸附CO2 的研究
CO2与CaO 的碳酸化反应是典型的气-固反应,第一步就是CO2分子在CaO 表面的吸附。早在1991 年,Cazorla 等[15]就通过TG-DTA、TPD 以及TPR 等手段对这重要的第一步进行了热力学和动力学研究,确认了在323−573 K 下CO2在CaO表面仅发生不可逆的化学吸附,CO2的吸附被限制在颗粒的表面。吸附(碳酸化反应)过程中,随温度升高,CO2的吸附覆盖率会增加,碳酸盐形成的越深入;碳酸化反应是气体体积减小的反应,增大反应压力可以提高反应的转化率;碳酸化反应的时间要适中,时间过短不能使反应进行完全,时间过长容易破坏吸附剂的孔结构。研究CO2在CaO 上的吸附行为,需要考虑外部质量传递、CO2孔内扩散、CO2扩散通过产品层以及化学反应这几个方面。在之后的研究中,对于CO2在CaO 上的吸附动力学、热力学以及微观吸附行为的研究不断增多。
1.1 吸附反应动力学和热力学
从宏观反应动力学上看,最早研究碳酸化反应动力学的是英国学者Barker[16],他系统研究了CaCO3⇄CaO+CO2可逆反应,发现CO2在CaO 上的吸附符合Ⅳ型等温线,认为CO2在CaO 上吸附分为两个阶段:化学反应控制的快速阶段和扩散控制的慢速阶段,之后的研究也证实了这一点[17−19]。在化学反应控制阶段,CO2与多孔CaO 材料表面的CaO 发生接触反应,随着反应的进行,CaO 颗粒被CO2碳酸化形成CaCO3产物层,之后的反应需CO2扩散通过CaCO3产物层,才能继续与内部的CaO 反应,转化率和反应速率下降。CaO 基吸附剂吸附CO2的动力学过程机理如图1 所示。
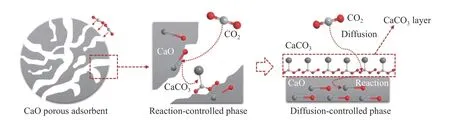
图1 CaO 吸附CO2 的碳酸化反应过程机理[16]Figure 1 Mechanism of carbonation reaction process of CaO with CO2[16]
在CaO 基吸附剂吸附CO2的动力学研究中,学者们建立了一些气固反应的动力学模型来对碳酸化过程进行描述,通过这些模型可以预测和计算碳酸化所需的活化能、产物层厚度、碳酸化反应速率和转化率等,可以更好地描述碳酸化反应过程,对CaO 基吸附材料的性能进行有效预测,也有利于工业过程碳酸化反应器的设计等实际应用。早在1980 年Bhatia 等[20]就建立了随机孔隙模型来描述碳酸化的动力学反应过程,除此之外还有收缩核模型[21]、晶粒模型[22]和速率方程理论[23]等,如表1 所示。

表1 碳酸化反应的动力学模型Table 1 Kinetic models of the carbonation reaction
收缩核模型从宏观上考虑了气固反应从外层向内层收缩的特征,体现了碳酸化的反应和扩散两个阶段,但是没有考虑到固体吸附剂内部的孔结构。晶粒模型在此基础上考虑了晶粒的体积膨胀、晶粒的形状因子等因素,同样没有考虑到吸附剂的孔隙结构对反应动力学的影响。而随机孔隙模型考虑了吸附材料内部的孔隙结构,用孔结构参数来表征动力学反应,更加符合碳酸化反应过程。这三种动力学模型较为简单易用,但都没有考虑到碳酸化过程中产物的成核和生长过程。之后Li 等[23]建立的速率方程理论对针对这一方面做出了改进,考虑了产物的成核和生成、表面反应及扩散、晶界和晶格扩散等方面,对碳酸化反应过程的描述更加准确。
从热力学上来看,CaO 晶体表面存在氧原子缺失导致的点缺陷、台阶边缘处的缺陷和角位缺陷等。Solis 等[24]通过微量量热法测量CaO 表面上CO2的吸附焓得出,CO2首先以单齿碳酸盐的形式吸附在台阶的边缘处,之后在台阶位点上吸附,吸附位点的稳定性从高到低为:单原子阶、拐角、边缘、双原子阶和平台。Besson 等[25]通过DFT理论计算从热力学角度对CaO 吸附CO2的吸附行为进行了分析,认为CO2在CaO 上的吸附遵循典型的Langmuir 行为,升高温度会降低CO2在CaO表面的覆盖率。低覆盖度(≤1/3 ML)时,CO2与CaO 间仅形成C−O 吸附键;中覆盖度(4/9−7/9 ML)时,CO2增多使部分Ca 原子和O 原子从表层离开,CaO 表面出现一些空隙;高覆盖度(≥ 8/9 ML)时,CO2可以从产生的空隙扩散渗透至表层以下,与下层的原子成键,使碳酸盐的形成逐渐深入,CO2在CaO 表面吸附的平均吸附能随覆盖度的增加呈先减小后增大的变化趋势[26,27]。
1.2 微观吸附
关于CO2在CaO 晶体上的吸附实验研究已多有报道,学者们主要通过热重分析等技术来研究,但在微观原子尺度上的分析受到了技术限制。近些年来,基于密度泛函理论(DFT)在理论模拟、计算材料学和计算化学等领域的广泛应用,一些学者开始利用该方法从原子尺度上对CO2在CaO 晶体上的吸附情况进行模拟计算,发现CO2在CaO 晶面上的吸附存在四种吸附模型[27,28]:氧顶位:CO2平行吸附在O 原子上;四重洞位:垂直吸附在Ca−Ca−O 空穴上方;二重桥位:垂直吸附在两个相邻Ca 原子间;Ca 顶位:垂直吸附在Ca 原子上。李晓东等[29]运用广义梯度密度泛函理论和周期平板模型方法,研究得出CO2最稳定的吸附位点为CaO(100)表面的氧顶位。四种吸附模型如图2 所示。

图2 CO2 在CaO 表面上的四种吸附模型[28]Figure 2 Four adsorption models of CO2 on CaO surface[28]
1.3 CaO 基吸附剂捕集CO2 的应用研究
学者们在CaO 基吸附剂用于燃烧后捕集CO2的基础研究方面开展了大量工作,但由于低成本、高效能的抗烧结CaO 基吸附剂制备工艺开发困难等原因,应用研究的步伐较为缓慢。日本学者Shimizu 等[10]最先开发了固体输送管线连接的“双流化床反应器钙循环捕集CO2”的工艺,即构建碳酸化反应器和煅烧再生反应器两个流化床反应器。之后的研究基本都是在该工艺的基础上发展的。双流化床系统循环捕集CO2的具体流程见图3。即CaO 材料在碳酸化过程稳定吸附CO2,CaCO3在煅烧过程中释放出CO2并重新得到CaO进入循环,过程中需排出煅烧反应器中因磨损和烧结等原因而失活的吸附剂,并补充新鲜的CaO吸附剂。在碳酸化反应器中CaO 需过量,确保CaO 与CO2发生化学吸附,否则CaO 不足将形成亚稳态的碳酸盐,在进入煅烧炉前可能就会分解为CaO 和CO2,影响吸附效率。
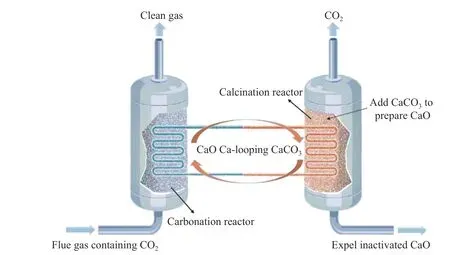
图3 CaO 循环捕集CO2 示意图[30]Figure 3 Schematic diagram of CaO recycling for CO2 capture[30]
基于双流化床钙循环捕集CO2的工艺,西班牙、德国等学者开始了中试应用研究。西班牙的Abanades 等[31]于2004 年搭建了一台小型中试流化床反应器,用CaO 从模拟的烟道气中吸附-脱附CO2,通过实验结果和模拟结果证明了双流化床钙循环捕集CO2是一种非常有效的CO2捕集的装置。2013 年,Arias 等[32]运行了1.7 MWth(热功率单位,全称为Megawatt Thermal)的钙循环捕集CO2中试工厂,在CFB 碳化器下捕集效率均超过90%,为该技术的实际应用提供了重要基础数据。同年,德国的Kremer 等[33]在达姆施塔特工业大学1 MWth钙循环捕集CO2试验工厂中进行了连续捕集CO2的测试,该试验工厂达到了90%的捕集效率。
CaO 基吸附剂燃烧后捕集CO2的技术从实验室规模的反应装置到工业化应用模拟、中试工厂运行和半工业化吸附剂制备工艺的研究,正在不断的发展,学者预测到2027 年将可实现钙基吸附剂循环捕集CO2的商业化运行[34]。在中国,2009年清华大学的蔡宁生课题组[35]建立了双鼓泡流化床反应器,可以长期稳定、连续地从烟气中捕集CO2,捕集效率可达95%,证明了CaO 基吸附剂捕集CO2大规模工业化应用的可行性。2011 年,李英杰[36]采用Aspen Plius 10.2 模拟软件将捕集系统分为煤气化、燃气、钙循环捕集CO2和余热锅炉及汽水循环四个子系统进行模拟,计算了氧碳比、气碳比、捕集效率等对系统净化率的影响,对钙循环的工业化应用起到了参考作用。2014 年,中国台湾的Chang 等[37]对台湾工业技术研究院于2012−2013 年建立运行的1.9 MWth的钙循环捕集CO2工艺的中试工厂进行了测试,发现该系统可稳定的从水泥厂烟道气中捕集CO2,效率保持在85%以上,该中试工厂被认为是未来建立示范工厂的里程碑。2019 年华中科技大学的刘文强课题组[38]研发了一种基于半工业化生产和实际应用的CaO 基吸附剂制备方法,即通过湿法混合将Ca12Al14O33掺入CaO 中,再通过喷雾干燥技术制备出混合均匀的改性吸附剂,最后通过挤出-滚圆法得到Al 稳定的CaO 基吸附剂颗粒,该工艺制得的吸附剂颗粒循环吸附性能良好,满足实际工业应用的需求,为钙循环工艺的工业化应用提供了支持。不同规模钙循环捕集CO2装置的运行结果证明了此项技术大规模工业化运行的可行性,未来将逐步建立CaO 碳酸化/煅烧循环捕集CO2的试点工厂、示范工厂、商业工厂,逐渐实现大型排放源的CO2减排。
从CaO 吸附CO2的化学反应,到反应动力学、动力学模型研究,以及微观吸附情况的模拟计算,再到实际的工艺应用,CaO 基吸附剂吸附CO2的理论知识和实际应用情况在不断的发展。CaO 基吸附剂用于燃烧后循环捕集CO2的技术难点在于循环捕集过程中因烧结、磨损等因素所引起的失活问题。
2 CaO 基吸附剂的烧结失活
吸附剂随着碳酸化/煅烧循环次数的增加会有失活现象,表现为CaO 转化为CaCO3的转化率下降或CaO 对CO2的吸附容量下降。导致转化率下降或吸附容量下降的原因主要有循环过程中吸附剂的磨损、硫中毒以及煅烧过程中吸附剂的烧结。吸附剂的磨损将造成吸附剂孔结构的破坏,从而导致CO2吸附容量下降,该因素主要通过掺杂耐磨损的掺杂剂来强化其机械强度而延长吸附剂寿命。硫中毒是由于烟气中存在SO2而产生的一种吸附剂中毒现象。SO2的酸性强于CO2,会优先与CaO 发生反应,生成高热稳定性的CaSO4,不会在煅烧脱附过程中分解为CaO,导致吸附剂失活[39,40]。在实际应用中,提高烟气脱硫的效率有利于减弱因硫中毒而引起的失活现象。烧结失活是吸附剂吸附CO2后再经过煅烧脱附以恢复其吸附能力过程中产生的一种失活现象,主要是由于CaCO3的分解温度高于CaO 的烧结温度而造成的,因此,烧结是不可避免的,烧结失活是造成CaO 基吸附剂失活最重要的因素之一。
2.1 CaO 基吸附剂烧结失活机理
CaO 失活使吸附剂吸附CO2能力显著下降的原因主要是孔结构变化导致的比表面积下降。Abanades 等[41]总结了CaO 基吸附剂随循环次数增加,捕集CO2能力下降的半经验公式:XN=(1−fw)+fw。其中,XN为循环N 次后吸附剂的碳酸化转化率,fm和 fw为常数,CaO 前驱体为石灰石时,fm=0.77,fw=0.17。CaO 吸附剂的烧结通常发生在明显低于CaO 熔点的温度,其机理[42,43]为:当固体达到塔曼温度时,离子的空穴扩散被激活,CaO 相邻晶粒之间接触增长,使晶粒结合并团聚长大,一些孔结构坍塌甚至消失,最终导致孔隙率下降和比表面积减小,使CaO 基吸附剂在新的循环中捕集CO2的能力下降。CaO 烧结失活机理图如图4 所示。
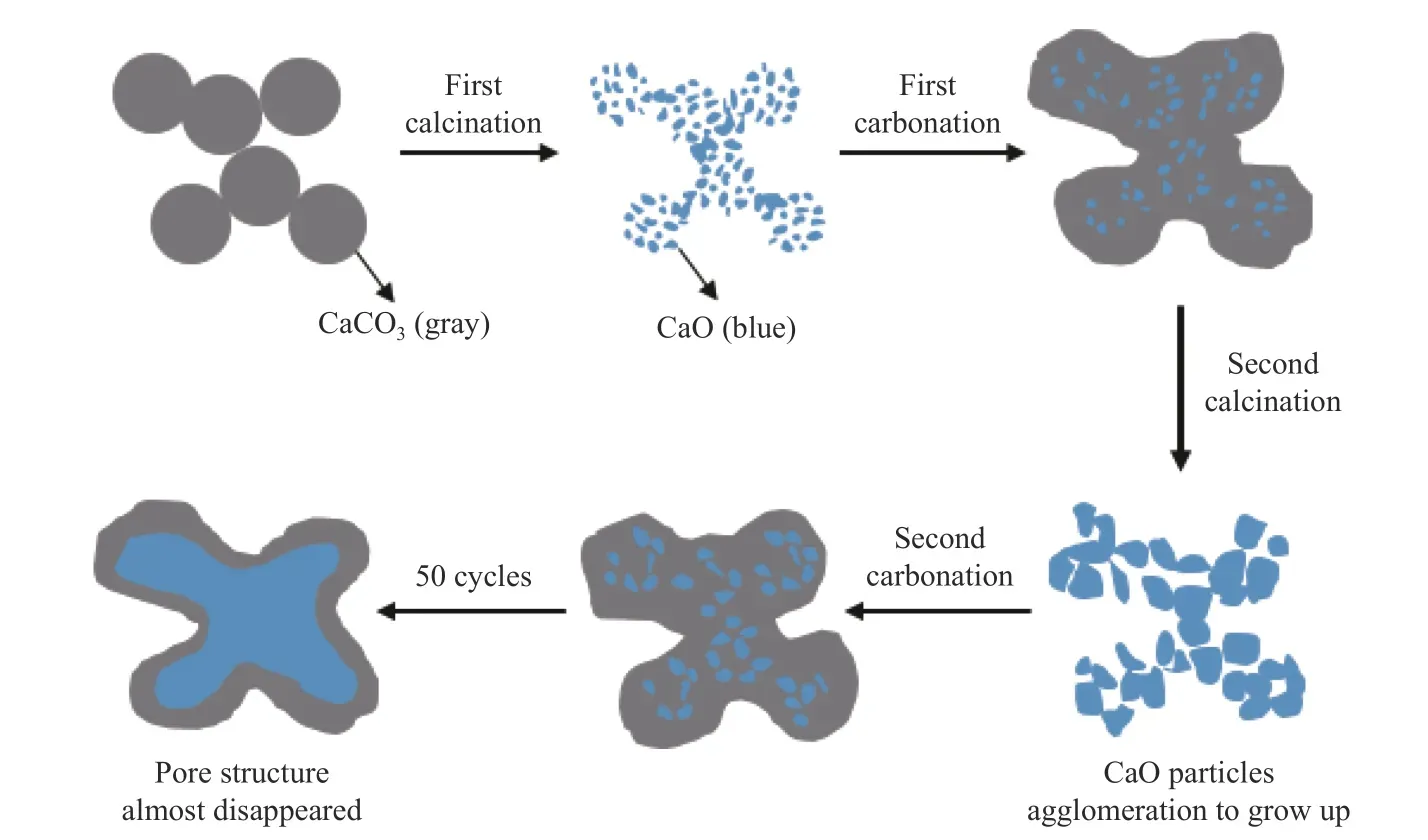
图4 CaO 烧结失活机理[44]Figure 4 CaO sintering deactivation mechanism[44]
近年来,Sun 等[45]研究了钙循环捕集CO2过程中孔结构的变化,发现在碳酸化/煅烧循环过程中,直径<220 nm 的孔结构收缩,>220 nm 的孔生长。其中<220 nm 的孔结构收缩是固态烧结的基本特征,而>220 nm 的孔生长是CaO 基吸附剂烧结的特有现象。小孔收缩、大孔生长导致了吸附剂比表面积的下降,使化学吸附反应转化率下降,吸附CO2容量降低。Bazaikin 等[46]研究了CaO 基吸附剂的烧结过程,认为烧结发生时CaO 晶粒间首先出现点接触,然后由于传质导致了晶粒之间“颈”的生长,最后由表面能梯度引起传质,导致晶粒聚集并减小了吸附剂的总表面积。烧结会导致多孔吸附剂孔隙率下降、比表面积严重损失,是CaO 基吸附剂最重要的影响因素之一,CaO 基吸附剂本身、碳酸化/煅烧循环条件都会对吸附剂的烧结产生影响。
2.2 吸附剂烧结的影响因素
温度高于771℃时CaO 会发生烧结,而CaCO3的分解温度达到825℃。在保证达到CaCO3分解温度的情况下,煅烧温度越高、煅烧时间越长、吸附剂粒径越大,吸附剂失活的情况越严重。图5总结了文献中不同煅烧时间和温度[30]、不同粒径大小[47]等影响因素下CaO 基吸附剂首次、5 次、15 次、20 次循环的碳酸化转化率。从图中可以看出,煅烧温度对吸附剂烧结的影响较大,Zhu 等[48]对不同煅烧温度下吸附剂的孔结构变化进行了研究,发现在750℃煅烧时中孔数量减少,微孔数量略有增加;850℃下煅烧两种类型的孔数量都显著减少;900℃煅烧时,在更短的时间内微孔数量就急剧减少。这表明煅烧温度越高,孔结构破坏越严重、比表面积衰减越快,烧结失活越严重。粒径大小会影响吸附剂的烧结,相对大粒径的吸附剂,随碳酸化/煅烧循环的增加,小粒径的吸附剂失活更慢。主要原因在于小颗粒的比表面积大多由外表面提供,缓冲了因烧结而引起的孔结构堵塞对总比表面积下降的影响,且有利于CO2扩散通过CaCO3产物层,使吸附剂活性下降速率减慢[47]。煅烧时间也会影响烧结,煅烧时间越长,吸附剂孔隙率越低,相较于煅烧温度,煅烧时间对吸附剂烧结失活的影响不大。除粒径、煅烧温度和时间外,钙前驱体也会造成吸附剂的烧结差异。Liu 等[49]研究了不同钙前驱体烧结失活的影响,结果表明,以D-葡萄糖酸钙制备的CaO 基吸附剂抗烧结能力最强,而以碳酸钙为前驱体制备的吸附剂抗烧结能力最差。吸附剂之间的差异是由于CaCO3成核速率不同导致的,以D-葡萄糖酸钙制备的CaO基吸附剂诱导期更短,碳化30 min 后转化率最高,抗烧结能力最强;而以碳酸钙为前驱体制备的吸附剂由于碳化过程中小孔的封闭更严重,抗烧结能力最差。

图5 不同影响因素下CaO 基吸附剂的烧结失活情况[30,47,49]Figure 5 Sintering deactivation of CaO-based adsorbent under different influence factors[30,47,49]
然而,吸附剂并非在每个循环中都表现为失活,在某些条件下吸附剂经过多次循环会产生碳酸化转化率随循环次数增加而增加的现象,不全程随循环次数的增加而下降,这是CaO 基吸附剂的自活化现象[50]。研究者们认为,CaO 碳酸化过程中形成的CaO 硬骨架起到了支撑作用,减缓了吸附剂的烧结、磨损和粉化,在之后的煅烧阶段,CO2的释放和CaCO3转化为CaO 的体积变化,使吸附剂层中出现许多微孔,增加了材料的比表面积,当这一阶段比表面积的增加大于因烧结、磨损引起的失活的损失时,在下一个循环中转化率和反应速率增加,发生自活化现象[51]。
3 CaO 基吸附剂抗烧结改性研究
上文表明,影响吸附剂活性衰减速率的因素主要有吸附剂的粒径、碳酸化/煅烧循环捕集过程中的时间和温度等。导致烧结的根本原因是晶粒的黏连,因而CaO 晶粒间的距离直接影响到CaO基吸附剂的烧结状况。因此,学者们通常采用掺杂的方法分离CaO 晶粒,扩大颗粒间的距离而缓解黏连和烧结。增大吸附剂的比表面积、构造更多的孔隙,从而提高吸附剂的吸附能力也成为重要的手段。此外,当粒径小至纳米级别后,吸附剂的比表面积主要由外部提供,内部孔结构的改变对比表面积的影响不大,在很多抗烧结改性的研究中,制备纳米级的吸附剂取得了良好的效果。抗烧结改性的结果基本均是在维持吸附剂的高孔隙率和大的比表面积的条件下实现的。基于以上思路,目前抗烧结改性主要有水合作用改性、酸溶液改性以及掺杂改性,主要围绕改善吸附剂结构、定向构造物相、优化制备工艺和技术等,旨在提高吸附剂对CO2的吸附能力以及抑制CaO 晶粒的烧结长大。此外,CaO 基吸附剂的抗烧结改性需要同时兼顾吸附剂的抗磨损、粉化以及CaCO3产品层覆盖等几个方面,以确保其循环稳定性、机械稳定性以及CO2的吸附容量不损失。
3.1 水合作用改性
在碳酸化和煅烧过程中通入水蒸气,在CaO基吸附剂循环捕集CO2的过程中进行水合作用处理,可以使因烧结而失活的 CaO 基吸附剂重新活化。
水合作用既可以作用于碳酸化反应阶段,也可以作用于煅烧阶段,均可对吸附剂的抗烧结性能起到积极的作用。水蒸气作用于碳酸化过程,提高CaO 基吸附剂碳酸化转化率的原理主要有以下几个方面[52,53]:
第一,使CaO 与CO2反应的系统中生成Ca(OH)2,其吸附能力优于CaO。
第二,Ca(OH)2与CO2反应的过程中会释放出水蒸气,丰富了材料表面的孔隙
第三,Ca(OH)2结构不稳定,在反应过程中容易崩塌、裂纹,使CaO 基吸附材料具有更大的比表面积。
煅烧过程中的烧结问题主要是由于CaCO3的分解温度高于CaO 的烧结温度,降低CaCO3的分解温度可以有效地减缓CaO 的烧结失活。从反应平衡角度来看,在煅烧阶段通入水蒸气,降低了煅烧反应过程中的CO2分压,使CaCO3转化为CaO的过程加快,既可以降低分解温度又缩短了分解停留时间[54],减缓了煅烧过程中的烧结。
在之后的研究中发现,对于水合改性,使用醇溶液处理CaO 基吸附剂会得到比水蒸气更好的效果。Li 等[55]对比了不同浓度水蒸气、乙醇/水溶液改性后吸附剂首次、5 次、10 次、15 次循环的碳酸化转化率(图6),可以看出加入乙醇后改性吸附剂的碳酸化转化率大于纯CaO 基吸附剂和纯水改性的吸附剂,并且溶液中乙醇的浓度越高,改性吸附剂的性能越好。这可能是因为乙醇分子的加入增强了水分子对氧化钙的亲和力和渗透力,使煅烧后的CaO 孔隙更丰富。

图6 水蒸气、醇溶液改性CaO 基吸附剂的碳酸化转化率对比[52,55]Figure 6 Comparison of the carbonation conversion of water vapor and alcohol solution modified CaO-based adsorbent[52,55]
水合作用改性操作简便、成本不高,虽然可以有效提高CaO 基吸附剂的碳酸化转化率、减缓烧结失活,但是经纯水和醇溶液改性的CaO 基吸附剂机械稳定性会所有降低,在循环捕集CO2的过程中容易因磨损和粉化而引起损耗。因此,如何改善水合作用改性过程吸附剂的机械稳定性成为其攻关的方向。
3.2 酸溶液改性
酸溶液改性主要是用乙酸、丙酸等有机酸溶液对石灰石进行预处理。由于酸溶液的加入,在煅烧石灰石制备CaO 时会得到孔隙率更丰富、比表面积更大的吸附剂[56]。加入酸溶液之后,在煅烧钙前体制备CaO 时会产生更多的气体。Hu 等[57]研究了甲酸、乙酸、丙酸、柠檬酸水合物、草酸二水合物、乳酸、L-(-)-苹果酸和L-(+)-酒石酸这几种有机酸对石灰石改性的性能,发现几种有机酸改性后吸附剂的碳酸化循环转化率排序为:L-(+)-酒石酸>丙酸>L-(-)-苹果酸>乙酸>水合柠檬酸>乳酸>草酸二水合物>未进行酸处理的吸附剂。使用酸溶液改性促进吸附剂抗失活性能的原因是在改性吸附剂制备过程中,酸化可以将CaCO3转化为有机钙,在煅烧分解过程中酸化的有机钙分解为CaO 时释放出更多的C、CO、H2O 等小分子物质,使孔隙结构更加丰富。不同酸溶液改性的吸附剂分子量、分解过程中释放的气体不同,各改性后CaO 基吸附剂的结构有所不同,这是造成改性吸附剂性能差异的主要原因。
酸溶液改性的最大问题是成本较高,乙酸的价格在2600−3000 元/吨,丙酸的价格在8000 元/吨左右,其他有机酸的价格也比较昂贵,低成本酸改性成为发展方向。一些采用工业废酸等成本较低的有机酸溶液对CaO 基吸附剂进行改性的研究逐渐受到关注。Sun 等[58]采用焦木酸废液中的丙酸对CaO 吸附剂进行改性,煅烧后,酸化的石灰石2−20 nm 的孔径分布增多,比表面积更大,碳酸化转化率更高、速率更快。使用焦木酸改性后的石灰石在100 个循环之后的碳酸化转化率为0.31,而使用纯乙酸溶液改性的石灰石经100 次循环后的转化率为0.4[56]。理论上,采用丙酸改性的石灰石性能应优于乙酸改性,采用废酸中的酸溶液对石灰石改性性能相较于纯有机酸会有所下降。因此,在采用酸溶液进行改性时需平衡成本和性能的矛盾。
3.3 掺杂改性
掺杂改性涉及对复合材料结构和性能的调控,工艺比较灵活,通过掺杂改性的吸附剂抗烧结性能较好,是CaO 基吸附剂抗烧结改性中比较热门的研究方向。对于在CaO 基吸附剂中掺杂引入的物质,有些物质会以本来的物相形态均匀分散在CaO 基吸附剂中(如MgO 等),有些物质会与CaO 发生反应(如Al2O3等),与CaO 化学键合形成钙基复合材料,使惰性材料的分散更加均匀,起到抗失活作用。从引入的物质作用于抗失活性能的原理来看,一些物质通过增强CaO 对CO2的吸附来提高吸附剂的抗烧结能力(如Na+),一些物质则作为骨架,通过阻止CaO 晶粒的迁移和生长来提高其抗烧结能力(如MgO、Al2O3等具有高塔曼温度的氧化物)。
3.3.1 掺杂含氧空穴的物质
在CaO 吸附剂中掺杂含氧空穴的添加剂如CeO2、ZrO2等可以减缓CaO 颗粒的团聚、提高CaO 对CO2的吸附,吸附能力的提高主要是由于氧空穴物质的掺杂促进了CO2通过CaO 吸附剂的逐层扩散。促进作用的原理是:存在的氧空穴可作为氧供体提供O2−,与 CO2形成再次分解后形成 CO2,并释放出 O2−回到原来占位,下一占位上的O2−与上一占位释放出的CO2形成,然后继续分解成 CO2,这样CO2可以一层一层的穿过 CaCO3层[59,60]。掺杂Ce、Zr 等氧化物改性后的吸附剂,表面孔隙更加丰富,孔隙率更高,经多次循环后颗粒团聚情况减轻,烧结情况减缓。Yoon 等[61]通过固相物理混合和柠檬酸溶胶-凝胶法两种方法制备了掺杂ZrO2的CaO 复合材料,通过物理混合法制得的复合吸附剂循环稳定性提高了约1.5 倍,溶胶-凝胶法制得的吸附剂则提高了13 倍。通过溶胶-凝胶法制得的吸附剂其中的Zr 与CaO 以化学键合的方式结合在一起,分散更加均匀,且溶胶-凝胶法使吸附剂颗粒尺寸减小,增强了其吸附动力学。
掺杂含氧空穴的物质,虽然会对碳酸化过程的扩散控制阶段产生积极的影响,但是掺杂的Ce、Zr 等这类富含氧空穴的物质价格较为昂贵,制约了大规模工业化应用。
3.3.2 掺杂钾、钠盐类
同样作用于碳酸化反应的扩散阶段而提高吸附能的掺杂剂还有钾、钠盐等,图7 总结了掺杂KCl、Na2CO3改性吸附剂循环过程的碳酸化转化率,可以看出掺杂0.5% KCl、Na2CO3后吸附剂的性能显著提高[62]。钾、钠盐掺杂改性的作用原理是通过增加碳酸化反应过程中CaCO3产物层的缺陷浓度,来增加Ca2+扩散通过产物层的通过率,同时可以降低解吸温度,基于表面反应提高CO2的吸附动力学,使CaO 在长期循环中保持稳定。Lee等[63]研究了Na2CO3掺杂对CaO 基吸附剂吸附CO2动力学的影响,制备的吸附剂生成了Na2Ca(CO3)2复盐,对CO2的亲和力大幅度提升,在第一个循环中表现出13.3 mol/kg 的较高吸附能力,并在七个循环之后稳定在6.5 mol/kg。在添加此类掺杂物质时,改变了吸附剂碳酸化反应的途径,改善了产物层的扩散动力学,形成的复盐使吸附剂更容易再生,从而提高了吸附剂的循环稳定性。掺杂Na2CO3对CaO 基吸附剂性能提升的机理如图8所示。

图7 掺杂钾、钠盐改性CaO 基吸附剂的碳酸化转化率对比[62]Figure 7 Comparison of the carbonation conversion of modified CaO-based adsorbents doped with potassium and sodium salts[62]
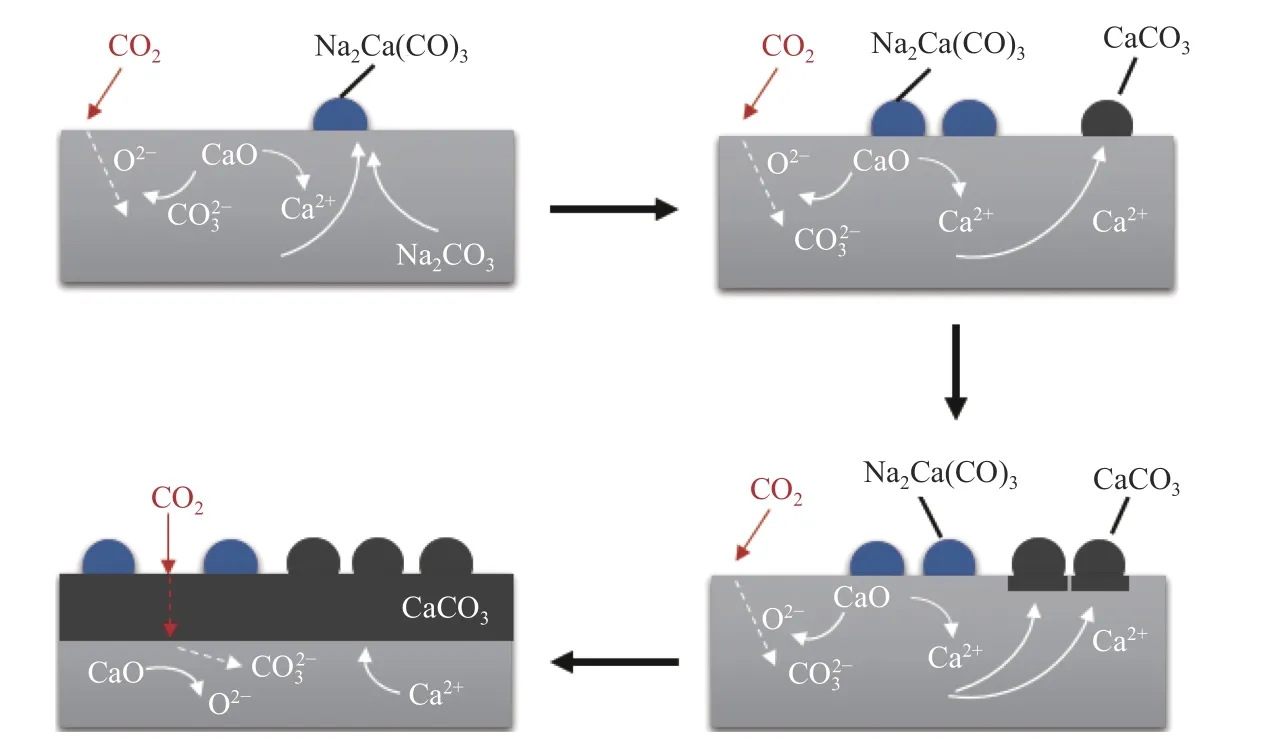
图8 掺杂Na2CO3 的吸附剂吸附CO2 机理[63]Figure 8 Mechanism of CO2 adsorption by Na2CO3 doped adsorbent[63]
近期研究显示,多孔CaO 比 CaCO3更适合作为浸渍掺杂钾、钠盐类的基质。Xu 等[64]采用同时水合浸渍法将钠盐掺杂在多孔CaO 中,发现改性吸附剂的大孔结构在循环过程中相对稳定,改性的效果提高了近一倍。同时,研究结果表明,掺杂的钠盐越多,吸附剂的稳定性越高,但过量掺杂钠盐会导致吸附剂初始循环容量较低。这是掺杂钾、钠盐类改性方法共存的问题。改性吸附剂表面形貌比较光滑,经BET 测得的比较面积低,虽然在扩散阶段增大了CO2扩散通过CaCO3产物层的通过率,但是在反应控制阶段,由于比表面积较低,会对CaO 吸附CO2的化学反应产生不利的影响。
3.3.3 掺杂塔曼温度高的惰性物质
还有一类掺杂剂,这类掺杂剂具有较高的塔曼温度,可以起到骨架作用阻止CaO 晶粒的团聚生长,有效的提高吸附剂的抗失活能力。这类掺杂剂主要是Al[65−69]、Mg[70]、La[71]、Ti[72]、Si[73]等的惰性氧化物。
掺杂高熔点的惰性氧化物如Al2O3、MgO 等,可以通过物理分离防止CaO 晶粒团聚,其机理如图9 所示。Liu 等[74]从原子角度上研究了此类氧化物抑制烧结的机理,发现CO2与Al2O3等氧化物发生表面相互作用时,电荷从表面转移到了CO2,这在能量上是有利的,且掺杂剂表面对簇的高吸附能可以限制CaO 团簇的迁移,使其不易烧结失活。Al2O3是常用的金属氧化物掺杂剂,掺杂Al2O3时会与CaO 反应生成Ca12Al14O33、Ca9Al6O18等惰性物质,均匀分布在复合吸附剂中。现在对于掺杂Al2O3的复合吸附剂主要是对其结构和形貌的调控,纳米中孔微球结构是吸附和循环稳定性能比较好的一种复合结构。Liu 等[66]采用硬模板法制备了掺杂Al 的CaO 纳米中空微球,其中空结构使碳酸化/煅烧循环过程中的局部大体积变化得到了缓冲,且形成的Ca12Al14O33起到物理屏障作用,防止聚集烧结,制得的复合吸附剂性能良好。在此基础上,Ma 等[75]采用生物模板(纸渣中的纤维、碳化物渣)制备了CaO/Ca12Al14O33中孔微管状复合吸附剂,由于生物模板的加入,复合吸附剂的孔隙结构更加丰富,且合成的材料易于去除、无残留。

图9 掺杂惰性组分抗烧结示意图[76]Figure 9 Schematic diagram of anti-sintering by doping with inert components[76]
引入其他惰性氧化物可分别作用于不同的场合、起到不同抗失活的作用。如用于增加甲烷重整制氢过程CO2捕集时,可掺杂Ni 的氧化物得到吸附-催化复合材料[77],既有利于催化过程,又可起到惰性材料支撑作用。掺杂两性金属氧化物TiO2可提高复合吸附剂的抗磨损能力[72],而掺杂SiO2则一般可将该惰性组分作为载体,利用SiO2独特的结构来减少孔堵塞[78],形成的Ca2SiO4也是良好的惰性骨架。有时也掺杂多种氧化物及调控复合材料的结构,可以达到比掺杂单种惰性材料更好的效果。如Peng 等[79]制备了核为Al2O3,外层为CaO,中间掺杂TiO2的核壳型复合材料,TiO2的掺入抑制了CaO 与Al2O3颗粒之间的反应,有效提高了复合吸附剂的反应活性,其循环稳定性明显优于纯CaO 和单独掺杂Al2O3的吸附剂。张明明等[80]同时掺杂了Al2O3和MgO,合成出CaOCa3Al2O6-MgO 的三元复合材料,MgO 的掺入提高了吸附剂的吸附容量,同时与Al2O3一起作为惰性骨架有效抑制了CaO 晶粒的烧结,效果优于单种掺杂剂改性。研究多种掺杂剂掺杂的复合吸附剂制备,可以平衡各掺杂剂单独加入的负面作用,得到性能更好的掺杂型CaO 基吸附剂。
掺杂惰性物质改性的CaO 基吸附剂,惰性物质分散在CaO 吸附剂中,碳酸化/煅烧循环之后,改性后的吸附剂保持了比纯CaO 更好的孔隙结构,惰性物质分散的越均匀,改性性能更好。将文献中掺杂的各惰性氧化物的改性吸附剂捕集CO2的性能总结于表2。

表2 掺杂各氧化物的改性吸附剂捕集CO2 的性能Table 2 CO2 capture performance of modified adsorbents doped with various oxides
从文献中可以总结出,掺杂氧化物对CaO 基吸附剂抗失活性能的改善能力从大到小为:Al>Mg≈La>Ti≈Si。在制备方法中,同等条件下采用溶胶-凝胶法制得的吸附剂可以获得极高的CO2捕集能力,这是由于该方法制备的改性吸附剂微观结构非常蓬松,颗粒排布更加致密细小,促进了高温吸附过程中吸附剂与CO2分子的结合及CO2的扩散[81]。采用粒径小的纳米材料或空心材料可以为吸附反应提供更大的比表面积,且表面积由材料外部提供,吸附性能和循环稳定性更好。通过掺杂氧化物改性的CaO 基吸附剂在多次循环中保持活性,有良好的循环稳定性,但是掺杂氧化物之后吸附剂的初始吸附容量大多有所下降。掺杂不同的氧化物、不同的制备方法以及不同的掺杂比例都会对吸附剂的性能产生不同的影响,在掺杂惰性氧化物改性的研究中,作者需要寻找合适的掺杂剂和工艺,并对抗失活改性复合材料的物相和结构进行定向调控,以及平衡吸附容量和循环稳定性两个方面的性能和降低掺杂改性所需的成本。
3.4 固废基抗烧结CaO 吸附剂
工业固废(如钢渣、赤泥、电石渣等)中含有大量的活性CaO,还富含Al2O3、SiO2等可作为掺杂剂的惰性物质,以工业固废为原料制备CaO 基CO2吸附剂不仅可降低CO2捕集的成本,还可同时实现CaO 基吸附剂的抗烧结改性,该方面的研究逐渐引起研究者们的关注。Liu 等[82]以电石渣为前驱体,通过鼓泡法合成了抗烧结性能良好的CaO 基吸附剂,材料中吸附活性组分为CaO(61.95%),惰性掺杂组分为Al2O3(3.44%)、SiO2(3.38%)和MgO(0.12%),最佳工艺条件下首次循环吸附量可达619.8 mg/g,并在多个循环内保持稳定。Tian 等[83]通过钢渣的结构重整,促进了钢渣中各物质捕集CO2的协同作用,其中,CaO 作为化学吸附CO2的活性物质,MgO 作为孔隔离剂以增加吸附剂的孔隙率,Al2O3作为稳定剂,抑制吸附剂的烧结。He 等[84]以鸡蛋壳为钙前体,掺杂了10%的赤泥,合成了以Ca12Al14O33为骨架的抗烧结钙基吸附剂,在100 个循环后依然保持了46.67%的优异转化率。粉煤灰等含大量惰性Al2O3、SiO2等物质的工业固体废弃物,也可作为理想的掺杂改性剂,降低改性成本[85−88]。将不同固废制备抗烧结CaO 基吸附剂的研究总结于表3,选用最佳工艺条件下合成改性吸附剂的循环捕集性能进行对比。

表3 固废基抗烧结CaO 吸附剂捕集CO2 的性能Table 3 CO2 capture performance of solid-waste-derived anti-sintering CaO adsorbents
与天然矿石相比,含钙固废的粒径更加细小,且来源地靠近CO2排放源,可以省去原料运输费用,有望进一步降低成本,具有商业化应用的潜质。固废基CaO 吸附剂的制备将成为重要的发展方向。但固废中CaO 的活性需要进一步激发,且需要与其中的Al2O3、SiO2等惰性组分协同,制备成具有更高循环稳定性、更高吸附容量的复合吸附剂。此外,固体废弃物中还含有大量Fe、Ti 等其他组分,这些组分的影响及其与CaO 的相互作用必将对固废基CaO 基吸附剂的性能产生重要影响。相关方面的研究将成为固废基CaO 基吸附剂开发的焦点。
4 结论
CaO 基吸附剂是捕集CO2的理想材料,应用于捕集CO2的理论研究、工业化进程正在不断推进,CaO 基吸附剂在碳酸化/煅烧过程中因烧结而失活的问题是制约该技术发展的瓶颈。
CaO 基吸附剂在碳酸化/煅烧循环捕集CO2过程中因CaO 晶粒团聚长大、孔结构坍塌导致吸附剂的孔隙率下降、比表面积减小,造成吸附剂的烧结失活,使吸附容量急剧下降。抗烧结改性可以维持吸附剂良好的孔隙结构以及大的比表面积,提高CaO 基吸附剂对CO2的吸附。抗烧结改性的研究主要包括水合作用改性、酸溶液改性和掺杂改性三种。水合作用改性可以使CaO 吸附剂在碳酸化反应阶段崩塌裂纹,获得更大的比表面积,在煅烧阶段降低反应温度和停留时间,减缓烧结;酸溶液改性的吸附剂制备时会产生更多的气体和小分子物质,孔隙率提高;掺杂改性可以促进CaO对CO2的吸附和扩散,还可以作为骨架分离CaO颗粒,阻止其迁移和扩散。其中,掺杂改性工艺灵活、改性后性能良好,是比较有前景的改性方法。多种掺杂剂协同作用可以实现各性能的互补,制得吸附性能、循环稳定性更好的复合吸附剂。
电石渣、钢渣等固体废弃物中含有大量的活性CaO 以及Al2O3、SiO2、MgO 等惰性物质,其中的CaO 可作为化学吸附CO2的活性物质,Al2O3、SiO2、MgO 等惰性物质可作为惰性掺杂剂提高抗烧结性能,利用含钙固废制备抗烧结CaO 基吸附剂技术上可行,并有效降低了捕集CO2的成本,将成为重要的研究方向。
