长双歧杆菌C16 bif黏附基因敲除菌株的构建
2021-08-11陈晓倩付恒芳朱旭萌刘丽波
谭 颖,陈晓倩,付恒芳,朱旭萌,李 春,刘丽波
(东北农业大学乳品科学教育部重点实验室 哈尔滨 150030)
双歧杆菌在人肠道中发挥重要作用,与人体的正常代谢及健康密切相关。而黏附是长双歧杆菌定植于肠道发挥作用的前提[1-7]。深入研究长双歧杆菌的黏附能力尤为重要,只有弄清其黏附机制,才能改善在人体肠道的定植状况。
目前报道的双歧杆菌黏附基因有BopA、Serpin、slpA、bif 等。Guglielmetti 等[8]从双歧杆菌MIMBb75 中分离纯化出对caco-2 具有黏附作用的BopA 蛋白。Jakava-Viljanen 等[9]从双歧杆菌ATCC8287 中筛选出slpA 表面黏附蛋白。Schell等[10]筛选出与黏附相关的Serpin 黏附蛋白。bif 黏附蛋白是Fujiwara 等[11]在长双歧杆菌SBT2928 中发现的,并证明bif 黏附蛋白不仅具有黏附作用,而且对致病菌的黏附能力有抑制作用,其对产志贺氏菌毒素大肠杆菌的抑制作用达到50%。bif 基因总长为1 059 bp,经NCBI 数据库分析其为长双歧杆菌NCC2705 编号58012118 的序列[12]。
本试验旨在构建长双歧杆菌bif 基因缺失菌株,验证长双歧杆菌bif 黏附基因的黏附性能。较为常见的基因敲除方法为插入失活、RNA 干扰以及同源重组[13-15]。孙金威等[16]利用同源重组方式成功敲除了保加利亚乳杆菌的ccpA 基因序列。本试验以同源重组方式敲除长双歧杆菌bif 黏附基因,为进一步研究长双歧杆菌bif 基因的黏附作用奠定基础。
1 材料与方法
1.1 材料与试剂
长双歧杆菌C16,实验室自备菌株;感受态细胞大肠杆菌(E.coli)DH5α,维地生物;质粒pMD19-T simple Vector、质粒PUC18,北京宝易生物有限公司;细菌基因组DNA 提取试剂盒、质粒小量提取试剂盒、琼脂糖凝胶DNA 回收试剂盒,北京索莱宝科技有限公司;DNA 纯化回收试剂盒,北京天根生化科技有限公司;2×Taq Mater Mix、2×Fast Pfu Mater Mix、λ/Hind Ⅲ DNA Marker、DL5000 DNA Marker、DNA ladder2000 Marker、T4DNA 连接酶,北京宝易生物有限公司;限制性内切酶EcoRⅠ、NotⅠ、PstⅠ,北京宝易生物有限公司;四环素、氨苄青霉素,Amresco 公司。
1.2 设备与仪器
SPX-150B 生化培养箱,上海一恒有限公司;HIRAYAMA HVE-50 型高压灭菌锅,HIRAYAMA公司;GL-21M 离心机,上海化工机械厂;AF100型制冰机,Scotsman 公司;恒温振荡培养箱、水浴锅、9700 型PCR 扩增仪,Applied Biosystems 公司;BA300 型数码显微镜,Motic 公司;ΜVP 凝胶成像系统仪,ΜVP 公司;电穿孔仪、200 mm 电转杯,Bio-Rad 公司。
1.3 试验方法

表1 引物序列Table 1 Primer sequences
1.3.1 目的基因的提取 采用天根试剂盒提取长双歧杆菌C16 的基因组,以提取的基因组为模版扩增bif 黏附基因,PCR 反应体系:2×Taq 25 μL,上、下游引物2 μL,目的基因模板2 μL,添加ddH2O 至50 μL;PCR 反应程序(94 ℃预变性90 s,98 ℃变性20 s,61 ℃退火20 s,72 ℃延伸60 s,循环30 次,72 ℃终延伸5 min,4 ℃保温)[12]。电泳检测成功后扩增bif 基因的上、下游同源臂。以质粒PBR322 为模版对tet 基因进行扩增,PCR 反应体系:2×Pfu 25 μL,上、下游引物2 μL,目的基因模板2 μL,添加ddH2O 至50 μL;PCR 反应程序:94 ℃预变性90 s,94 ℃变性20 s,58 ℃退火20 s,72 ℃延伸60 s,循环30 次,72 ℃终延伸5 min,4℃保温;PCR 产物经1%琼脂糖凝胶电泳,切下目的片段,用琼脂糖凝胶DNA 回收试剂盒回收目的片段,利用2×Taq Mater Mix 进行3'端加A。
1.3.2 目的基因与T 载体的连接及纯化 将4 μL 纯化的PCR 产物与1 μL 载体pMD19-T simple Vector 放入EP 管中,加5 μL 缓冲液,4 ℃过夜连接,构建重组质粒pMD19-T simple Vectorup、pMD19-T simple Vector-down、pMD19-T simple Vector-tet。从-80 ℃冰箱中取出感受态细胞大肠杆菌DH5α,立即放在冰上缓化30 min,将连接好的重组质粒全量10 μL 分别导入100 μL感受态细胞大肠杆菌DH5α 中,置冰上30 min 后42 ℃热激75 s,再置冰上1 min,加入890 μL SOC培养基,150 r/min 振荡培养1.5 h,平板涂布,37 ℃培养12~16 h。挑取单菌落进行菌落PCR 验证,将阳性单菌落挑到质量浓度为100 μg/mL 的Amp液体培养基中培养12~16 h,用快速质粒小提试剂盒提取重组质粒,送样测序,将测序结果与标准菌株比对。按照限制性内切酶使用说明书方法用EcoRⅠ和NotⅠ对pMD19-T simple Vector-up 进行酶切,用NotⅠ对pMD19-T simple Vector-tet进行酶切,用NotⅠ和PstⅠ对pMD19-T simple Vector-down 进行酶切,用EcoRⅠ和PstⅠ对质粒PUC18 双酶切处理。
1.3.3 基因片段的连接及纯化 将up、down、tet和载体PUC18 的酶切纯化产物按照T4DNA 连接酶使用说明书连接(16 ℃连接6 h),构成敲除载体PUC18-up-tet-down,再将敲除载体转化到感受态细胞大肠杆菌DH5α 中培养(方法参照1.3.2 节)。菌落PCR 验证成功后,提取敲除载体,电泳检测提取产物,进行酶切纯化。以纯化产物为模版,以bif 基因序列的引物进行PCR 扩增,电泳检测PCR条带,切胶回收条带,将纯化的基因条带送样测序。将测序结果与标准序列比对,若测序结果正确,将未酶切的敲除载体导入感受态细胞大肠杆菌DH5α,验证敲除载体的稳定性。
1.3.4 敲除载体的转化
1)线性打靶片段的构建 以构建好的敲除片段为模版,以bif-F1 和bif-R1 为引物扩增线性打靶片段(PCR 反应体系:2×Fast Pfu 25 μL,模版1 μL,上游引物2 μL,下游引物2 μL,加ddH2O 至50 μL)。PCR 反应程序:94 ℃预变性90 s,98 ℃变性20 s,58 ℃退火20 s,72 ℃延伸30 s,循环30次,72 ℃终延伸5 min,4 ℃保温[17]。电泳检测PCR产物,后切胶回收。
2)感受态的制备 将长双歧杆菌涂布于平板上,37 ℃培养48 h,镜检单菌落。将确认无杂菌的长双歧杆菌传代培养至第2 代,将二代菌用含0.5%的甘氨酸的Tpy 培养基培养至吸光度为0.5 A,冰浴30 min 使菌体停止生长,取10 mL 菌液6 000 r/min 离心10 min,弃上清,加同体积的灭菌并冰浴的去离子水,6 000 r/min 离心10 min,重复上一步,用同体积的30%PEG-8000 悬浮菌体6 000 r/min 离心10 min,弃上清,再用200 μL 30%PEG-8000 悬浮菌体,后立即进行电转化[18]。
3)电转化 取2 μL 重组质粒和5 μL 线性打靶片段与200 μL 感受态细胞混合后转移到电转杯中,在2.5 kV/cm 电压下进行电转化,立即放入900 μL 复苏培养基(含有0.5 mol/L 蔗糖的Tpy 培养基)中,37 ℃孵育2 h。孵育完成后,5 000 r/min离心1 min,吸出800 μL 培养液,吸取剩余培养液100 μL 涂布于含有四环素(5 μg/mL)的固体培养基中,37 ℃培养48 h[19]。
4)基因缺失菌株筛选及验证 在平板上挑取单菌落作为菌落PCR 模板,电泳检测PCR 产物,平板涂布阳性克隆菌株,挑取单菌落,接种到5 mL Tpy 培养基中,37 ℃培养后提取基因组。以提取的基因组为模板,扩增打靶片段。用1%琼脂糖凝胶电泳检测。将上一步所得阳性菌落传代培养,以提取基因组DNA 为模版,分别用bif-F、tet-R 和tet-F、bif-R 为引物做交叉验证试验,电泳检测目的基因条带[16]。
1.3.5 技术路线图 见图1。

图1 试验技术路线图Fig.1 The experiment technology roadmap
2 结果与分析
2.1 长双歧杆菌DNA 的提取结果
选取分离自健康婴儿肠道内具有高黏附性的长双歧杆菌C16 为研究对象,用试剂盒提取长双歧杆菌C16 的DNA,如图2所示,用0.7%琼脂糖凝胶电泳检测,在23.1 kb 下方得到长双歧杆菌基因序列条带。由此表明按照细菌基因组DNA 试剂盒说明书操作可以提取出长双歧杆菌C16 的全基因组。

图2 细菌基因组DNAFig.2 Bacterial genomic DNA
2.2 基因片段纯化回收
图3电泳结果显示,在1 000 bp 处检测出清晰的目的基因条带,与bif 黏附基因大小基本相同,切胶回收bif 黏附基因片段,电泳检测回收条带并送样测序。将测序结果与bif 黏附基因标准序列对比,相似度为99.92%,由此表明长双歧杆菌C16 中含有bif 黏附基因。彭珍[12]以长双歧杆菌WBLO01 的DNA 为模版,扩增出bif 黏附基因。本试验根据GenBank 报道的长双歧杆菌bif 黏附基因序列设计特异性引物。扩增出目的基因条带的同时含有明显的杂带,可能是因遗传序列具有高度保守性,引物发生改变而使其特异性下降,导致引物与基因间发生非特异性结合,扩增出非目的基因片段,因此含有明显杂带。此结果与Ruiz-Villalba 等[20]研究结果一致,退火温度决定PCR 特异性及产量,退火温度低造成引物与模版错配,发生非特异性结合;退火温度高,特异性增强,而退火过高会影响引物与模版的结合,因此应选择合适的退火温度。将退火温度60 ℃提至61 ℃,成功扩增出单一的bif 黏附基因条带。

图3 目的基因PCR(a)及纯化(b)结果Fig.3 Target gene PCR(a) and purification(b) results
如图4所示,以扩增出的bif 黏附基因为模板,扩增其的上、下游同源臂,用1.5%琼脂糖凝胶电泳检测,在250 bp 上端发现清晰的目的基因条带。切胶纯化回收基因片段,为bif 黏附基因上、下游同源臂与t 载体pMD19-T simple Vector 连接做准备。

图4 目的基因上(a)、下(b)游PCR 纯化结果Fig.4 PCR purification results of the upstream(a)and downstream(b) target gene
如图5所示,在1 000 bp 上部发现清晰的基因条带,表明成功扩增出tet 抗性基因片段,切胶回收,为tet 抗性基因与t 载体pMD19-T simple Vector 连接做准备。限制性内切酶Not Ⅰ全部由碱基CG 组成,提高了引物中的CG 的含量。当GC含量在70%以上时,用普通PCR 条件难以扩增出目的基因片段。因为G、C 之间形成3 对氢键,解链所需能量较高,所以模板较难打开,并且单链的GC 丰富区容易自身互补配对形成引物二聚体,不能与模版结合,DNA 聚合酶也难于延伸或停止延伸,出现严重的非特异性条带,甚至扩增不出靶基因[21]。本试验在引物两端加入保护碱基序列来降低引物中的GC 含量,参照2×Fast Pfu PCR 试剂盒说明书将变性温度调至94 ℃,退火温度调至58℃,成功扩增出正确的tet 抗性基因片段。利用2×Taq Mater Mix 扩增出3' 端含有A 的tet 抗性基因片段。

图5 tet 抗性基因PCR(a)及纯化(b)结果Fig.5 Resistance gene PCR(a)and the purification(b) results
2.3 基因片段的连接及酶切
如图6所示,bif 黏附基因上、下游同源臂和tet 抗性基因片段成功连接到t 载体pMD19-T simple Vector 上,分别形成重组质粒pMD19-Tup、pMD19-T-down、pMD19-T-tet。采用pMD19-T通用引物测序。将测序报告与bif 黏附基因上、下游同源臂和tet 抗性基因比对,结果显示:bif 黏附基因上、下游同源臂与标准序列相似度都为100%,tet 抗性基因与标准序列相似度为99.88%。本试验选取t 载体pMD19-T simple Vector 的原因是pMD19-T simple Vector 上包括EcoRⅠ、NotⅠ和PstⅠ在内的限制性内切酶位点均被消除,若连接在t 载体上的片段是bif 黏附基因上、下游同源臂和tet 抗性基因片段,则保证重组质粒上只会含有一个特异性酶切位点,连接失败,限制性内切酶无法切开重组质粒[22]。
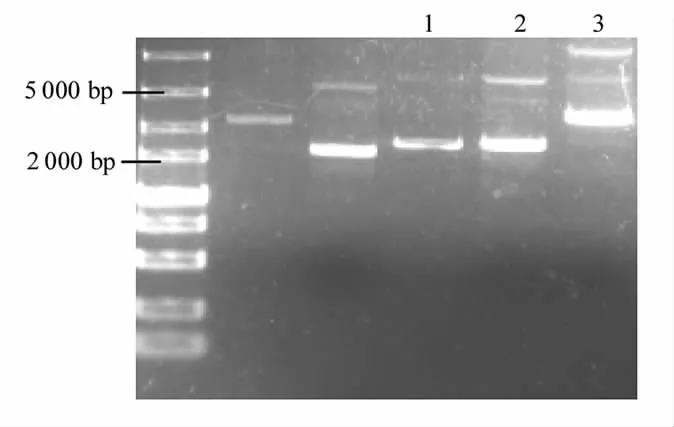
图6 bif 粘附基因上、下游及抗性基因连接结果Fig.6 Upstream and downstream and resistance gene junction results of bif adhesion gene
如图7和图8所示,利用限制性内切酶EcoRⅠ、NotⅠ和PstⅠ成功从3 种重组质粒上切下bif黏附基因,基因上、下游同源臂及tet 抗性基因片段。遵从Gawin 等[23]的酶切位点设计原则(切位点选择的基本原则是酶切位点单一,基因内部不能含有酶切位点,酶切效率高以及所有限制性内切酶公用buffer)选取EcoRⅠ、NotⅠ及PstⅠ为本试验的限制性内切酶。在bif 黏附基因上游同源臂两端加入EcoRⅠ和NotⅠ,在下游两端加入NotⅠ和PstⅠ,在tet 抗性基因两端加入NotⅠ。限制性内切酶标准用量是0.1 μL 切100 ng 的DNA。用量过低无法切开重组质粒,用量过高会将重组质粒切成若干片段。酶切体系中的甘油含量控制在5%,其过高导致限制性内切酶失活,影响酶切效率。本试验改进了传统方法,即提高限制性内切酶用量至0.12 μL,切100 ng DNA,用超纯水替代洗脱缓冲液作为重组质粒溶剂,成功切开重组质粒。

图7 bif 粘附基因上(a)、下(b)游质粒酶切结果Fig.7 The digestion results of upstream(a)and downstream(b) plasmid of bif adhesion gene

图8 tet 抗性基因质粒酶切结果Fig.8 The digestion results of plasmid of tet resistance gene
2.4 敲除载体的构建
图9表明通过T4DNA 连接酶成功将bif 黏附基因上、下游同源臂和tet 抗性基因连接到载体PUC18 上构成敲除载体PUC18-up-tet-down。采用通用引物对敲除载体测序,测序报告与敲除片段up-tet-down 标准序列相似度为99.78%。再次将构建成功的敲除载体PUC18-up-tet-down 转化到感受态细胞大肠杆菌DH5α 中,重复上述步骤,检测敲除载体的稳定性。此结果与万翠香等[24]的研究结果一致。
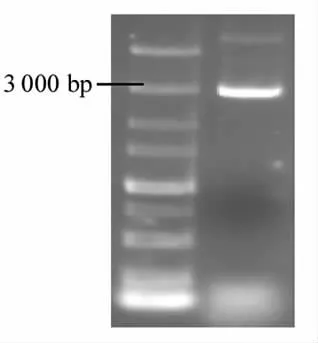
图9 重组质粒质粒连接结果Fig.9 The ligation results of recombinant plasmid
2.5 基因缺失菌株鉴定结果
挑取电转化后培养成的单菌落进行菌落PCR鉴定,引物为bif-F 和bif-R,若发生同源交换,则扩增出长度为1 800 bp 的片段;若未能发生同源交换,则扩增出长度为1 059 bp 的片段。如图10所示,1 号泳道成功扩增出1 800 bp 的片段。2 号泳道同时扩增出1 800 bp 和1 059 bp 的片段。3号以及4 号泳道只扩增出1 059 bp 的片段。决定电转化成功率的一大关键因素是细胞弱化剂。长双歧杆菌是革兰氏阳性菌,细胞壁较厚,本试验用甘氨酸为细胞壁弱化剂,将细胞壁肽桥之间的丙氨酸置换,阻碍细胞壁的修复与合成,在细胞壁上形成孔洞,提高电转化的成功率[25]。决定电转化成功率的另一因素是电压,电压太低导致细胞壁难以形成通道,敲除载体难以进入细胞内部;电压太高会击穿菌体细胞,导致菌体死亡,影响菌体生长。达到长双歧杆菌最佳电转效率的电场强度为2.5 kV/cm[18]。3 号和4 号泳道说明该宿主细菌的细胞壁上因甘氨酸处理不彻底而导致无法形成通道,敲除载体和打靶片段难以进入宿主细胞内部。1 号泳道说明打靶片段与bif 黏附基因成功完成同源交换,而2 号泳道说明虽然敲除载体和打靶片段进入宿主细胞内部,但未完成同源交换,这是因为遗传序列有高度的保守性,打靶片段上虽有bif 黏附基因的同源序列,但不能完全保证其能整合到宿主基因上,由此表明通过同源重组的方法将打靶片段整合到宿主基因上的成功率并不是100%,存在一定的概率性[26]。

图10 菌落PCR 验证结果Fig.10 PCR verification results of colony
对扩增出正确片段的1 号泳道对应的菌落用含tet 抗生素(10 ng/μL)的Tpy 培养基扩大培养,提取DNA 进行PCR 验证。图11电泳结果显示:在2 000 bp 下方成功扩增出长度约1 800 bp 基因条带,切胶回收片段,测序报告与打靶片段标准序列相似度为99.88%。

图11 bif 突变菌株PCR 验证结果Fig.11 PCR results of bif mutant strain
2.6 PCR 验证
如图12所示,利用两对引物分别成功扩增出长度为1 491 bp 的片段。本试验采用孙金威等[16]的PCR 交叉验证方式对基因缺失菌株进行验证,如果打靶片段整合到宿主DNA 上,则用bif-F 和tet-R 为引物扩增出长度为1 491 bp 的基因片段。此结果与孙金威等[16]研究结果一致,说明打靶片段up-tet-down 成功整合到宿主DNA 上。

图12 PCR 交叉验证Fig.12 PCR cross validation
3 结论
本试验成功扩增出长双歧杆菌C16 的bif 黏附基因,以此为基础成功扩增出该菌bif 黏附基因的上、下游同源臂,重组质粒pMD19-T simple Vector-up、pMD19-T simple Vector-down、pMD19-T simple Vector-tet。在此基础上酶切出上、下游同源臂及tet 抗性基因,并成功将上、下游同源臂及tet 抗性基因片段连接到载体PUC18 上,利用电转化的方式将敲除载体PUC18-up-tet-down 以及打靶片段up-tet-down 导入感受态长双歧杆菌中,实现其bif 黏附基因的敲除,为进一步研究长双歧杆菌bif 基因的黏附作用奠定基础。
