先天性中枢性低通气综合征6例病例系列报告
2021-08-11尹雅郡陈鸣艳马晓路
尹雅郡 谢 添 陈鸣艳 马晓路
1 病例资料
回顾性收集2017年3月至2021年2月在浙江大学医学院附属儿童医院(我院)住院确诊的6例先天性中枢性低通气综合征(CCHS)病例。CCHS诊断符合以下标准[1,2]:①存在肺泡低通气(低氧血症及高碳酸血症),并排除心肺及神经肌肉原发疾病;②基因检测证实存在PHOX2B基因突变。
从病历中采集患儿出生史、发病时间、临床表现、辅助检查、基因检测、治疗及最终结局。本研究为回顾性分析,经我院伦理委员会批准(批准文号:2021-IRB-020)并免除知情同意书。
表1显示,男4例、女2例,顺产和剖宫产各3例。6例均于新生儿期发病。例4有出生窒息史,当地医院予新生儿复苏抢救,Apgar评分不详,无缺氧缺血性脑病(HIE)临床表现。余5例无出生窒息史。父母均体健,例6的姐姐2年前在我院行剖腹探查及肠黏膜活检确诊全结肠型巨结肠,生后第2 d放弃治疗死亡(未行基因检测)。余5例否认家族遗传性疾病及类似疾病史。
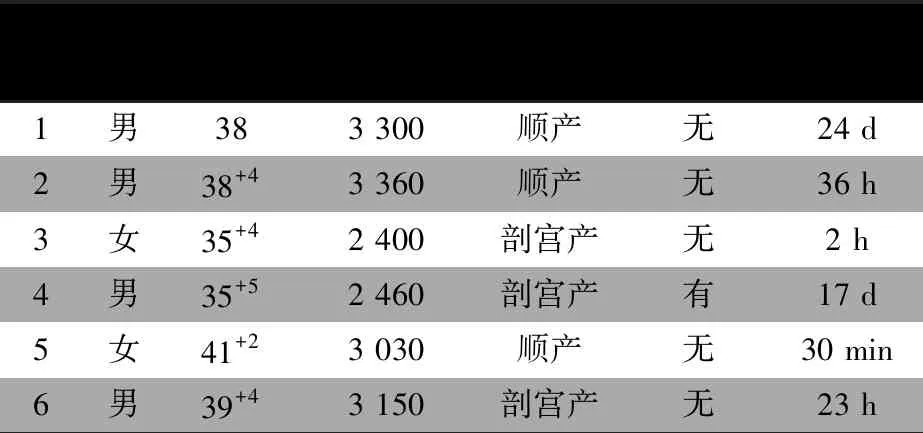
表1 CCHS患儿一般资料
表2显示,6例均表现为呼吸浅弱或自主呼吸少,反复发绀,例3~5睡眠时症状明显。均存在撤机困难,撤机后出现呼吸暂停,严重二氧化碳潴留(>100 mmHg)。例2入院后未曾撤机改无创辅助通气,入院前血气示二氧化碳分压102 mmHg。5例存在腹胀及喂养困难,其中例3和5有胎便排出延迟,例4和6经直肠黏膜活检/肠壁多点活检诊断先天性巨结肠(HD)。3例行脑脊液检测,脑脊液常规、生化均正常,脑脊液培养阴性。6例均行头颅B超检查,其中例5提示室管膜下出血不伴有脑室内扩散。5例行头颅MR检查,例1额叶点状异常信号影,例4枕部硬膜下少量出血,例5脑白质散在异常信号,考虑HIE。6例均行心脏超声检查,例1~3卵圆孔未闭,例4和5房间隔缺损,例6未见异常。6例甲状腺功能检测、遗传代谢筛查、X线胸片均未见异常。6例基因检测均示PHOX2B基因突变,5例为多聚丙氨酸重复序列突变(PARM),20/27基因型3例,20/26和 20/25基因型各1例,1例为非多聚丙氨酸重复序列突变(NPARM)。
表2显示,患儿入院后均予有创呼吸机辅助通气。例2未曾撤机,生后9 d放弃治疗后死亡。余5例患儿在呼吸机参数较低且生命体征稳定、血气分析正常时撤离有创呼吸机改无创辅助通气,但病程中均有撤机失败再次插管情况,其中例3生后84 d放弃治疗后死亡,例5自动出院后1个月死亡,例6生后29 d放弃治疗后死亡。例1于59日龄带无创呼吸机出院,出院后家中予双水平正压通气支持(BiPAP),半年后家长自行停用,现3岁5个月,生长发育良好,已读幼儿园。例4因HD行结肠造瘘术,5月龄带无创呼吸机出院,家中予BiPAP通气支持及结肠造瘘口护理,7月龄回我院行造瘘回纳手术,1岁时于睡眠中死亡。

表2 CCHS患儿临床资料
2 讨论
CCHS是一种以呼吸中枢的代谢控制障碍为特征的罕见病,在活产婴儿中的发病率为1/148 000~1/200 000[3,4],多于新生儿期发病,目前国外报道1 000余例,国内共报道40余例且多数预后不良[5]。临床主要表现为呼吸暂停、反复发绀、二氧化碳潴留和撤机困难等,症状典型的患儿清醒时通气良好,睡眠时通气不足。CCHS多在生后不久出现症状,但亦有儿童期或成人发病的报道,称为晚发型CCHS(LO-CCHS)[6,7]。部分患儿合并神经源性肿瘤、HD或其他自主神经功能障碍表现,如心律失常、晕厥、胃肠动力异常、体温调节障碍、多汗等[8,9]。Goldberg等[10]研究发现CCHS患儿存在眼部异常,如斜视、弱视、眼睑下垂及瞳孔缩小等。另有研究报道CCHS患儿有典型的面部特征,表现为脸部偏短和扁平[11]。但本文6例患儿并未发现有特殊面容。
CCHS诊断需排除原发性心肺疾病和神经肌肉系统疾病。2003年,Weese-Mayer团队[12]及Amiel等[13]的研究表明PHOX2B基因突变为CCHS的致病基因,PHOX2B基因突变包括PARM(基因型20/24-20/33)和NPARM(错义突变、无义突变、移码突变等),其中PARM占90%以上,较常见的基因型为20/25、20/26和20/27。本文6例PHOX2B基因突变中,5例为PARM,其中20/27基因型3例,20/26和20/25基因型各1例,另1例为NPARM。研究认为,PHOX2B基因型与表型之间存在相关性,PARM患儿丙氨酸重复延展越长,临床表型越重。20/24和20/25基因型很少需要24 h持续辅助通气,20/27-20/33基因型通常需持续通气支持且合并HD及其他自主神经系统症状的比例高,NPARM患儿临床症状更重,多数需持续通气支持[9,14]。本文临床症状相对轻且唯一存活的患儿为PARM20/25基因型,与此前研究结果相符。
目前CCHS无有效药物治疗,最主要的治疗方法是呼吸支持,包括气管切开正压通气、无创BiPAP辅助通气、负压通气及膈肌起搏。关于负压通气和膈肌起搏治疗的研究报道较少。美国胸科协会建议CCHS患儿首选气管切开正压通气,以保证有效的通气及最佳的氧合,改善神经系统预后,6~8岁时可考虑改无创辅助通气[1]。气管切开造口要选择合适大小气管套管,以避免气管软化、溃疡、肉芽肿形成等并发症,同时尽可能减少漏气的发生。新生儿及儿童患者建议使用不带囊的气管套管,定期评估,随着年龄体重的增长不定期更换合适大小气管套管。气管切开造口术的护理至关重要,包括切口皮肤的消毒护理、气管套管固定防止滑脱、防止异物堵塞造成窒息等[15]。气管切开正压通气需进行气体温湿化处理,防止痰栓形成,按需有效吸痰,减少过多的刺激以及气管黏膜的损伤。因气管切开术护理难度大,特别是CCHS患儿需长期在家中经气管造口呼吸机辅助通气,家长的接受度低,本文6例患儿亦因家属不能接受气管切开而未采取此治疗方案。BiPAP可在呼吸周期中提供吸气相和呼气相两个不同水平的压力支持,从而改善通气及氧合。BiPAP的优势在于无创伤性,不需要气管插管或气管切开,而是经鼻塞、鼻罩或面罩进行通气支持,且BiPAP呼吸机便携、价格相对便宜、容易操作。此外有研究认为BiPAP可成功应用于CCHS患儿的治疗[16-18]。本文2例患儿带无创呼吸机出院,家中予BiPAP辅助通气,1例于1岁时死亡,1例存活至今。BiPAP辅助通气可能出现漏气、鼻中隔或皮肤损伤、腹胀、呕吐及误吸等并发症,长期使用可造成面部发育不良,故需密切监测,使用时选择合适大小鼻塞或鼻面罩,腹胀明显患儿可予胃管减压。
研究表明,约20%的CCHS患儿合并HD[19],由于神经嵴起源的肠神经元发育异常,导致远端肠管痉挛狭窄,近端肠管扩张,临床表现为胎便排出延迟、腹胀、呕吐、便秘等,X线片可见肠管充气扩张、低位性肠梗阻。诊断方法包括钡剂灌肠造影、直肠肛管测压及直肠黏膜活检,治疗上可行肠造瘘术或根治术[20]。本文5例存在腹胀症状,其中2例胎便排出延迟,腹部X线片均提示肠管充气扩张明显,2例经活检病理证实HD,并行肠造瘘手术。
CCHS为终身性疾病,需患儿及其家长和医护人员共同合作,出院前医护人员应对家长进行培训和护理宣教,包括呼吸机、脉搏血氧饱和度仪和制氧机等仪器的使用,呼吸机应有蓄电功能,家中最好有备用呼吸机。应保证SPO2≥95%,根据患儿情况设置合适的报警值。家长应掌握基本的心肺复苏技能,如果有气管切开或肠造瘘,需掌握相应的护理技巧。CCHS患儿建议定期门诊随访,对呼吸状态进行评估,评价生长发育及神经认知发育情况。因病程中可能会出现反复低氧发作,所以应定期复查心电图、超声心动图及头颅MR,评估有无肺动脉高压、肺心病及缺氧缺血性脑病的发生。
