毛细管胶束电动色谱法同时分离检测补骨脂及其制剂中的5种活性成分
2021-08-09曹秋娥
易 婷,曹秋娥,李 菲
(云南大学 化学科学与工程学院,云南 昆明 650500)
补骨脂(Psoralea Fructus)是传统中药,属豆科补骨脂植物,又名破故纸、胡韭子等,表面呈黑色或棕褐色,主要生长在山坡、溪边等地,具有补肾壮阳、温脾止泻的功效,此外还有抗肿瘤[1-2]、抗抑郁[3]等作用,外用主要用来治疗白癜风等皮肤病[3].因此,对补骨脂中各活性成分研究的分析检测具有重要意义.目前已有近百种成分从补骨脂中分离得到,包括香豆素类、黄酮类、苯并呋喃类和单萜酚类等类型的化合物,其中香豆素类和黄酮类化合物的代表物质为补骨脂素、补骨脂甲素和补骨脂乙素等.
目前,已有文献报道对补骨脂中的香豆素和黄酮类成分进行分离分析研究,主要包括高效液相色谱法[4-7]、薄层色谱法[8]和高效毛细管电泳法[9-10],但已报道的方法都没有对补骨脂中补骨脂素、补骨脂甲素、补骨脂乙素、补骨脂定和补骨脂宁这5种活性成分进行同时分离检测的报道.毛细管电泳具有操作简单、分离效率高、样品用量少、维护运行成本低等优点,因此本文采用毛细管电泳法,建立了一个能同时分离检测这5 种活性成分的毛细管胶束电动色谱新方法,优化了缓冲溶液和仪器条件,在23 min 内实现5 种活性成分的有效分离和准确检测,可用于补骨脂药材及其复方制剂中这5种 活性成分的分析检测.
1 实验部分
1.1 仪器和材料P/ACETMMDQ 型高效毛细管电泳仪配有DAD 检测器(Sciex,美国);未涂层石英毛细管,57 cm×ϕ75 μm,有效长度50 cm(河北省永年锐沣色谱器件有限公司);pHS-2C 型pH 计(上海雷磁仪器厂).
药材补骨脂经云南大学生科院陆树刚鉴定为豆科植物补骨脂(Psoralea corylifoliaL.)的干燥成熟果实,鱼鳔补肾丸(成分:鱼鳔胶、枸杞子、莲须、肉苁蓉、巴戟天、杜仲、当归、菟丝子、补骨脂、茯苓、淫羊藿、肉桂、沙苑子、牛膝、附片)和复方补骨脂颗粒(主要成分:补骨脂、锁阳、续断、狗脊、赤芍、黄精)购于云南昆明福林堂药店;甲醇、乙腈、硼砂、氢氧化钠、十二烷基硫酸钠(分析纯,天津市风船化学试剂有限公司);补骨脂素(Psoralin,批号zl20150122)、补骨脂甲素(Coryfolin,批号zl20150312)、补骨脂乙素(Corylifolinin,批号zl20150411)、补骨脂定(Psoralidin,批号zl20150318)和补骨脂宁(Corylin,批号zl20150228)标准品(结构式见图1,购于南京泽朗医药科技有限公司);超纯 水(优普).
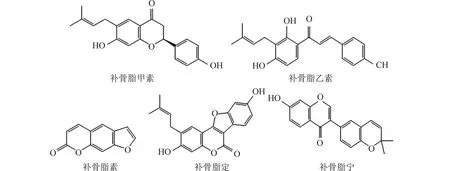
图1 5 种待测组分的结构式Fig.1 The structures of five investigated compounds
1.2 溶液配制
1.2.1 标准品溶液 质量浓度均为1.0 mg/mL 的补骨脂素、补骨脂甲素、补骨脂乙素、补骨脂定和补骨脂宁标准储备液由甲醇配制,不同质量浓度的标 准品工作溶液均由储备液用甲醇稀释得到.
1.2.2 样品溶液 于90 ℃真空干燥箱中烘干补骨脂药材、鱼鳔补肾丸和复方补骨脂颗粒,分别准确称取约1.000 0 g 烘干后的样品置于250 mL 锥形瓶,加入50 mL 体积分数为50%的甲醇溶液,59 kHZ 超声提取60 min,过滤,以体积比为50%的甲醇溶液洗涤残渣3 次,合并滤液和洗涤液,减压蒸 干,残渣用甲醇溶解并定容至25 mL.
1.2.3 运行缓冲溶液 以20 mmol/L Na2B4O7(用NaOH 调节pH 9.31)为缓冲溶液,加入体积分数为25%乙腈和25 mmol/LSDS 为添加剂.
以上溶液在使用前均用0.45 μm 微膜过滤处理;除运行缓冲溶液外,其余溶液均置于4 ℃冰箱中 避光保存.
1.3 实验方法以20 mmol/L Na2B4O7(用NaOH调节pH 9.31)为缓冲溶液,加入体积分数为25%乙腈和25 mmol/L SDS 为添加剂.仪器条件为:分离电压20 kV,毛细管柱温25 ℃,5 s 压力进样(3.448 kPa),检测波长300 nm.
仪器开机和重复运行之间,用0.1 mol/L 的NaOH 溶液、纯水和缓冲溶液依次冲洗毛细管柱4、3 min 和4 min.
2 结果与讨论
2.1 运行缓冲溶液的pH 值及浓度对分离的影响由图1 中5 种目标组分的化学结构可知,除补骨脂素外,其他4 种目标组分都含有酚羟基,在碱性介质中容易电离而带上负电荷.因此,实验选择了在碱性介质中进行电泳分离测定,在pH 值介于8.26~11.06,Na2B4O7浓度在15~35 mmol/L 的范围内研究了Na2B4O7(用NaOH 调节pH)缓冲溶液的pH 和浓度对待测组分分离的影响(图2).结果表明,pH 值和硼砂浓度越高,各组分的迁移时间逐渐增加.当pH 低于9.64 时,补骨脂甲素和补骨脂宁的电泳峰完全重叠在一起;当pH≥10.26 时,除补骨脂素外,其他4 种目标组分都实现了基线分离.当pH 达到11.06 时,除补骨脂素外的4 种组分都实现了有效分离与检测.但此条件下由于pH 过高导致电流过大,测定时容易断流,不利于分析检测的重现.所以,为了提高分离度,缩短分离时间,并改善基线噪音,实验选择以20 mmol/L 的Na2B4O7(用NaOH 调节pH=9.31)作为运行的缓冲溶液.
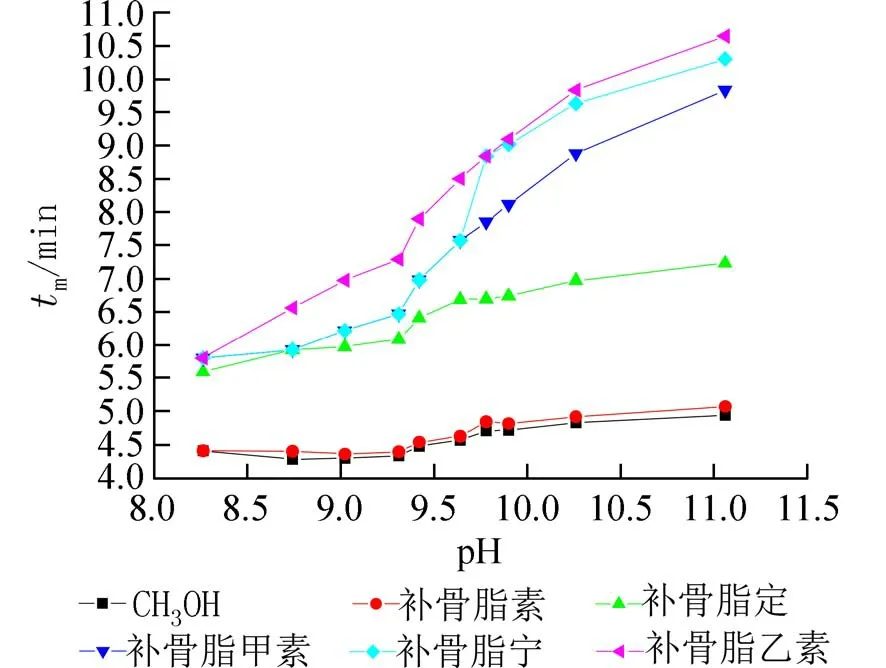
图2 缓冲溶液pH 值对各组分迁移时间的影响Fig.2 Effect of buffer solution pH on migration time of investigated compounds
图3 是在浓度为20 mmol/L 的Na2B4O7(用NaOH 调节pH 9.31)缓冲溶液中5 种标准品混合溶液的电泳图.可以看出,补骨脂甲素和补骨脂宁的电泳峰此条件下完全重合,并且甲醇和补骨脂素的 电泳峰也没有得到有效分离.
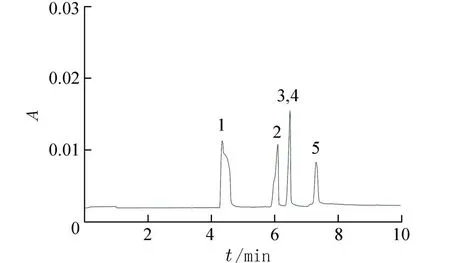
图3 浓 度 为20 mmol/L 的Na2B4O7(用NaOH 调 节pH 9.31)缓冲溶液中5 种标准品混合溶液的电泳谱图Fig.3 Electropherogram of the standard mixed solution in Na2B4O7(20 mmol/L)buffer solution(adjust pH to 9.31 with NaOH)
2.2 乙腈对分离的影响在浓度为20 mmol/L 的Na2B4O7(用NaOH 调节pH 9.31)中,补骨脂甲素和补骨脂宁的电泳峰完全重合,并且甲醇和补骨脂素电泳峰也重合在一起(图3).为了改善各组分间的分离,首先在运行缓冲溶液中加入乙腈作添加剂,在乙腈体积分数位于10%~30%的范围内,研究了对各组分分离度的改善情况(图4).结果表明,随着乙腈浓度的增加,各组分的迁移时间都有所延长,补骨脂甲素和补骨脂宁之间能够实现基线分离,但对于补骨脂素和溶剂甲醇之间的分离度没有明显改善.因此,实验选择在运行缓冲溶液中添加25%(体积分数)的乙腈来改善分离度,此条件下的标准品 混合液电泳图见图5.
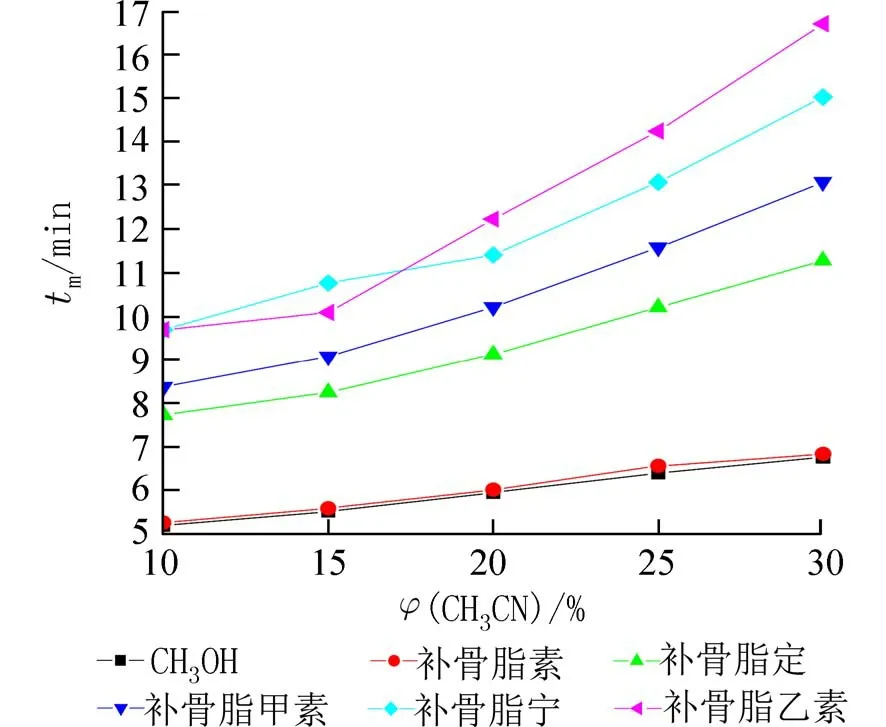
图4 乙腈体积分数对各组分迁移时间的影响Fig.4 Effect of acetonitrile volume fraction on migration time of investigated compounds
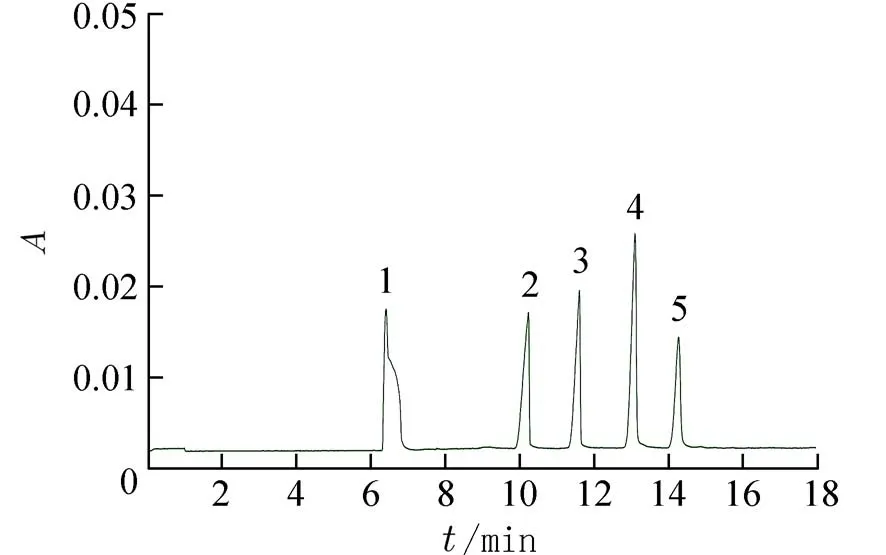
图5 在20 mmol/L Na2B4O7(用NaOH 调节pH 9.31)缓冲溶液中添加25%乙腈后的标准品混合溶液电泳图Fig.5 Electropherogram of the standard mixed solution in Na2B4O7(20 mmol/L)buffer solution(adjust pH to 9.31 with NaOH)containing 25% acetonitrile
2.3 十二烷基磺酸钠(SDS)浓度的影响由图5可见,在上述条件下,除补骨脂素的谱峰还和甲醇峰完全重叠在一起外,其余4 种组分之间都达到了基线分离.因此,为了改善补骨脂素和中性溶剂甲醇的分离度,在含有体积分数为25%的乙腈的20 mmol/L 的Na2B4O7(用NaOH 调节pH 9.31)缓冲溶液中加入SDS,采用胶束电动毛细管电泳模式来改善补骨脂素和甲醇的分离.研究了SDS 浓度在10~30 mmol/L 的范围内对各组分分离效果的影响(图6).结果表明,随着SDS 浓度的增加,各组分间的分离度逐渐增大,同时补骨脂素和甲醇电泳峰的分离度也有所提高.但当SDS 浓度小于25 mmol/L时,补骨脂素的电泳峰裂分情况严重,只有SDS浓度增大至25 mmol/L 时,补骨脂素的峰形相对较好.因此,考虑在运行缓冲溶液中再加入浓度为2 5 mmol/L 的SDS 以改善目标组分的分离度.
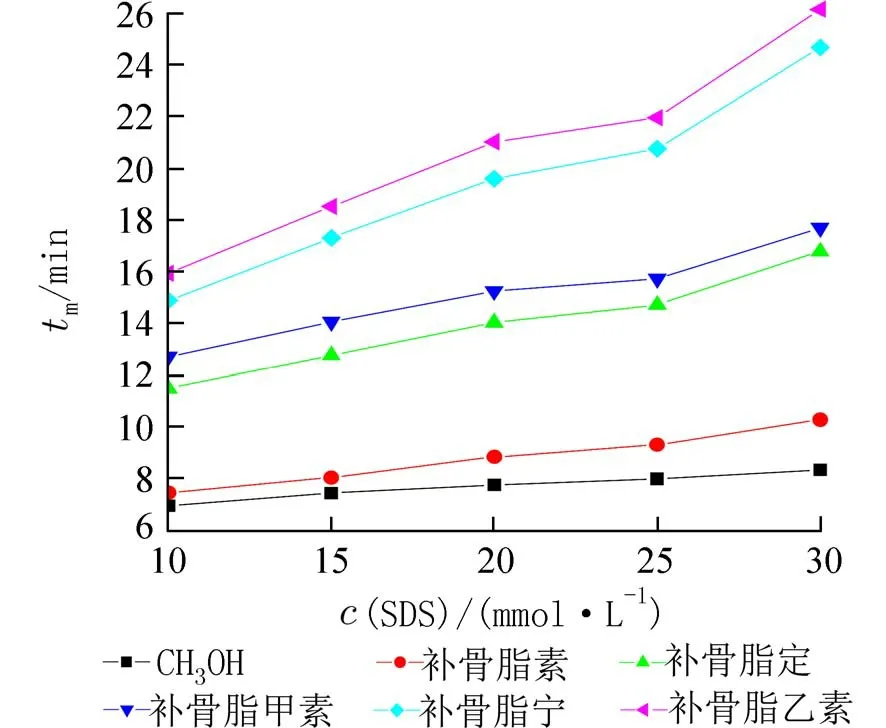
图6 SDS 浓度对各组分迁移时间的影响Fig.6 Effect of the SDS concentration on migration time of investigated compounds
2.4 仪器条件在优化了上述运行缓冲溶液的基础上,实验对分离电压和进样量等仪器条件进行了优化.结果表明,在15~28 kV 的范围内,增大分离电压,各组分的迁移时间会缩短,分离度下降且峰形变窄;在进样时间为3~9 s 的范围内逐渐增加,各组分的峰面积均增加且峰形展宽,降低了补骨脂素和溶剂甲醇之间的分离度.综上,实验最终选择了20 kV 的分离电压、3.448 kPa 压力进样5 s.
在上述优化后的电泳条件下,5 种活性成分的标 准品混合物电泳图见图7.

图7 5 种活性成分的标准品混合溶液的电泳图[缓冲溶液组成:含有25%(体积分数)乙腈和25 mmol/L SDS 的20 mmol/L 的Na2B4O7(用NaOH 调 节pH 9.31)缓冲溶液]Fig.7 Electropherogram of standard mixed solution of five active components
2.5 工作曲线在最终优化条件下,对方法的特征性进行了考察.将标准储备液稀释得到一系列不同质量浓度的5 种组分的标准品混合溶液,在最终优化条件下的运行缓冲溶液中进行电泳分离检测,以各组分的峰面积为纵坐标,质量浓度为横坐标绘制 了该方法的工作曲线,结果见表1.
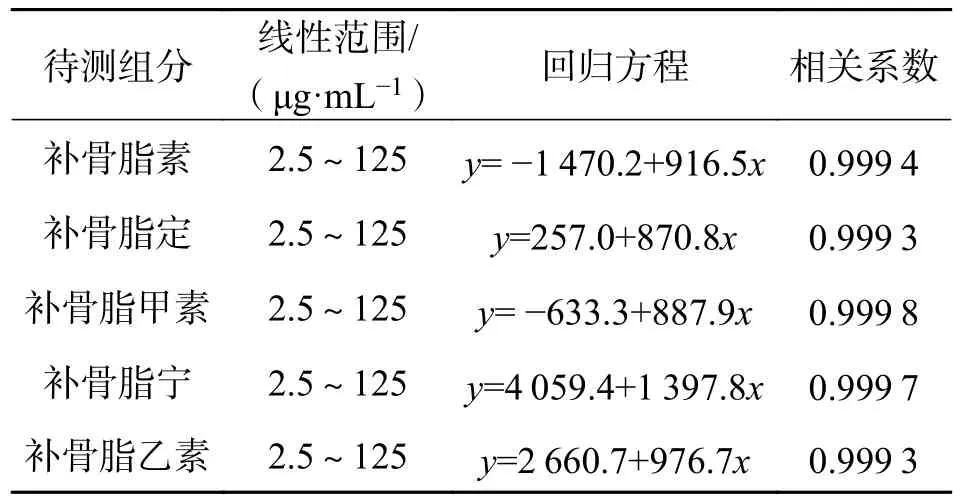
表1 待测组分的工作曲线和精密度Tab.1 Working curve and precision of investigated compounds
2.6 方法学验证
2.6.1 重复性验证 配制了6 份质量浓度相同的供试品5 种待测组分标准品的混合溶液(相当于100%浓度水平)进行测定,测定结果见表2.可见,6 次测定结果5 种待测组分的峰面积的RSD 值均小于5%,保留时间的RSD 值均小于3%,说明方法的 重复性符合要求.
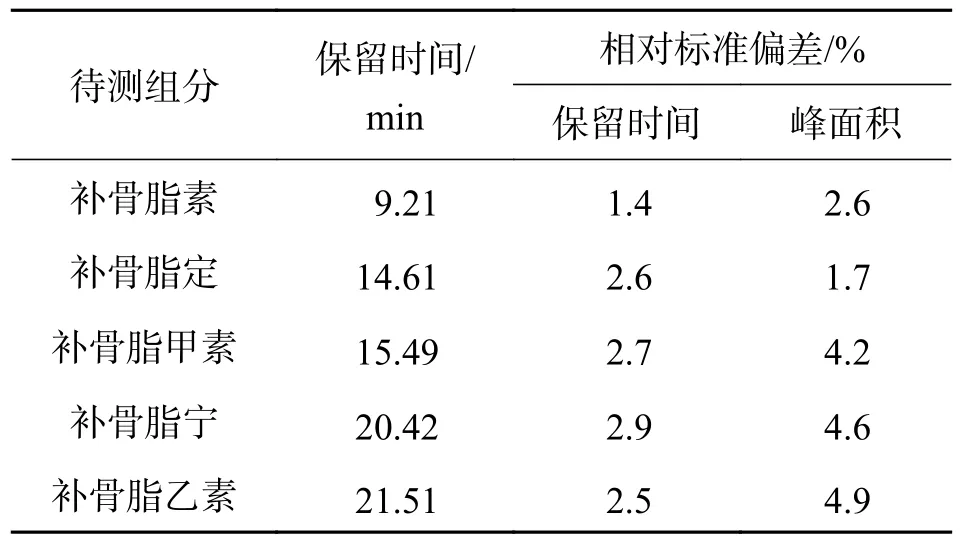
表2 重复性验证结果(n=6)Tab.2 The results of epeatability verification (n=6)
2.6.2 中间精密度验证 采用同一分析人员不同日期、不同分析人员对5 种待测组分标准品的混合溶液(质量浓度为25 μg/mL)进行了测定,验证方法的中间精密度,结果分别见表3 和表4.
由表3 可见,同一分析人员连续2 天,每天进样测定6 次,所得全部测定结果5 种待测组分标准品的混合溶液峰面积的RSD 值为1.8%~4.7%,保留时间的RSD 值为1.1%~3.1%.由表4 可见,两个不同分析人员,每人进样测定6 次,所得全部测定结果的峰面积的RSD 值为1.7%~4.3%,保留时间的RSD 值为1.3%~3.3%.上述结果表明,本方法 的精密度符合要求.
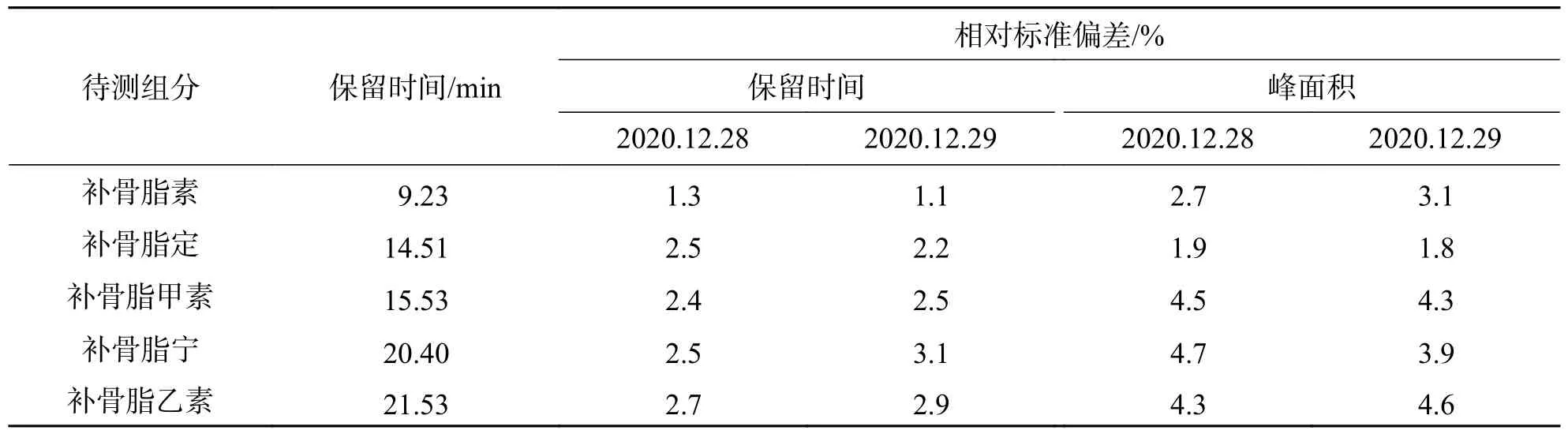
表3 中间精密度验证(同一人不同日期测定结果,n=6)Tab.3 Intermediate precision verification (n=6)

表4 中间精密度验证(不同分析人员测定结果,时间:2020.12.30)Tab.4 Intermediate precision verification(results determined by different analysts,December 30,2020)
2.6.3 稳定性验证 取同一份补骨脂药材样品,在0,2,4,6,8 h 分别进样以测定补骨脂素、补骨脂甲素、补骨脂乙素、补骨脂定和补骨脂宁的含量,结果见表5.可见,5 种待测组分的含量的相对标准偏 差均小于5.0%,表明方法的稳定性符合要求.

表5 稳定性验证结果(n=5)Tab.5 The results of stability verification(n=5)
2.6.4 准确度验证 以测定实际样品加标回收率验证方法的准确度.各补骨脂药材及其复方制剂的加标回收实验结果见表6.可见,5 种待测组分的加标回收率都在95.2%~104.8%之间,加标回收率的相对标准偏差均小于5.0%,表明方法的准确度符 合要求.

表6 样品中5 种目标成分回收率测定结果(n=3)Tab.6 The recoveries of five investigated compounds in samples(n=3)
2.6.5 最低检测限和定量限 将最低质量浓度的补骨脂素、补骨脂甲素、补骨脂乙素、补骨脂定和补骨脂宁对照品溶液分别进行逐步稀释,按上述所建立的方法进行检测.按照LOD=3.3δ/S(δ为标准曲线截距的标准偏差;S为标准曲线的斜率)计算了方法的检出限,按照LOD=10δ/S计算了方法的定量限,结果见表7.结果表明,方法的检测限和定 量限符合要求.

表7 最低检测限和定量限Tab.7 The results of limits of detection and quantification.
2.7 样品分析为了验证方法的实际应用价值,将上述所建立的方法用于1.2 的3 种样品溶液中5种目标组分的分离测定.各补骨脂药材及其复方制剂的测定结果见表8.补骨脂药材样品的电泳图见图8.测定结果说明本文建立的方法可以用于补骨脂 中这5 种组分的含量测定,具有一定的实用价值.
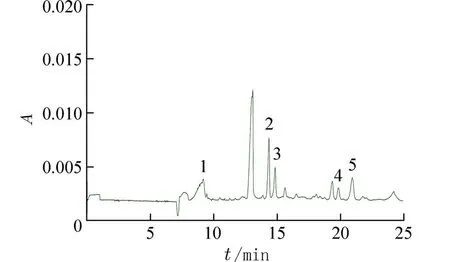
图8 中药材补骨脂样品的电泳图Fig.8 Electropherogram of Chinese traditional medicine psoralen
3 结论
本文采用毛细管胶束电动色谱分离模式,在详细优化了运行电解质溶液组成和仪器条件下,建立了一个能同时分离检测药材补骨脂及其制剂中补骨脂素、补骨脂甲素、补骨脂乙素、补骨脂定和补骨脂宁5 种活性成分的新方法.该方法可以在23 min内实现补骨脂药材及其复方制剂中这5 种目标组分基线分离与有效检测,具有较高的实用价值,为补骨脂及相关制剂中这些有效成分的定性和定量分析提供了新方法.
