重铬酸钾滴定法测定废杂铜中的铁含量
2021-08-05苗晓焕冯振华
苗晓焕 冯振华
(1.北矿检测技术有限公司,北京 102628;
前言
随着我国国民经济的持续快速发展,铜矿资源的短缺和精铜需求的快速增长之间的矛盾日益突出。废杂铜原料的再生利用具有能耗低、工艺相对简单、综合效益高等特点[1],可极大弥补铜资源的不足,进口废铜仍是目前中国再生铜行业的主要原料来源,约占三分之二左右[2]。其中含铜低于90%的中低品位废杂铜由于来源广泛,成分复杂其综合利用能力较薄弱[3],主要包括报废的铜废件、电子或机械加工过程中的废料、铜粉、铜渣、含铜烟尘、铜灰、废旧电线电缆等铜基合金[4]。
在利用废杂铜冶炼精炼铜过程中,铁对氧气的亲和力大,造渣性能好的特点使其能迅速且较完整地除去。但对含铁量>3%的杂铜而言,火法精炼时,铁先氧化形成氧化亚铁与石英形成硅酸盐炉渣[5],同时放出大量的热量使炉温急骤上升,从而造成炉子严重腐蚀的后果。另外在电解过程中,不同价态的铁离子会在阴阳两极来回氧化放电,降低电流效率[6]。因此,为了更好地指导冶炼生产,亟需建立一种适用于测定废杂铜中铁含量的检测方法。
废杂铜基体复杂,干扰因素众多,目前针对废杂铜中铁含量的测定没有相关的国家和行业标准方法,现行的《铜及铜合金化学分析方法》(GB/T 5121—2008)中铁含量的测定范围为0.0001%~0.0020%,而实际样品中如铜粉、铜渣中铁含量远超此范围,因此该方法对较高含量的铁不适用。
本文采用盐酸、硝酸、氢氟酸、高氯酸分解样品,氨水沉淀生成氢氧化铁与杂质元素分离,将其应用于废杂铜试样分析,准确度高,结果满意。
1 实验部分
1.1 主要试剂
盐酸、硝酸、氢氟酸、高氯酸、氯化铵、氨水、过氧化氢均为分析纯,实验用水为去离子水。
二氯化锡溶液:称取5 g二氯化锡于40 mL盐酸(1+1)加热溶解后,用水稀释至100 mL,混匀。
硫-磷混酸(15∶15∶70):将500 mL浓硫酸在搅拌下慢慢加入2 000 mL水中,冷却后加500 mL磷酸搅拌均匀。
重铬酸钾标准滴定溶液:称取4.389 7 g预先在140~150 ℃烘2 h的重铬酸钾(基准试剂),于500 mL烧杯中,加300 mL水溶解后移入2 000 mL容量瓶中,用水稀释至刻度,摇匀。此溶液1 mL相当于2.5 mg铁。
铁标准溶液的配制:称取2.50 g纯铁至500 mL锥形烧瓶中,慢慢加入35 mL盐酸(1+1),加热至溶解,冷却后少量多次加入5 mL过氧化氢氧化溶液,加热至沸,分解过剩的过氧化氢,移至1 000 mL容量瓶中,稀释至刻度。1.00 mL该溶液相当于1.00 mL的标准重铬酸钾溶液。
氯化铵-氨水洗液:25 g氯化铵溶于500 mL热水中,加20 mL氨水混匀。
三氯化钛溶液(1+14):移取2 mL三氯化钛溶液15%~20%(m/V)用盐酸(1+5)稀释至30 mL,现用现配。
钨酸钠溶液(250 g/L),二苯胺磺酸钠溶液(5 g/L)。
1.2 实验方法
称取3.00 g试样(分筛样品按筛分后的质量比合称,精确到0.000 1 g)于300 mL烧杯中,加少量水润湿试料,加入10 mL盐酸、15 mL硝酸,难溶样品滴加氢氟酸助溶。盖上表面皿,置于电热板低温处缓慢加热至反应停止。用滴管少量多次沿杯壁缓慢滴加5 mL硝酸,继续溶解至试料分解完全,将烧杯取下,稍冷后加入10 mL高氯酸,继续加热至冒白烟并回流4~5 min,用水吹洗表面皿和杯壁。待溶液充分冷却至室温后,转入250 mL容量瓶中(如有黑渣需过滤做补正),加水稀释至刻度,充分摇匀后静置。移取25.00 mL溶液于300 mL烧杯中,加水至75 mL,向溶液中加入4~5 g氯化铵固体,加热至氯化铵固体完全溶解且溶液微沸,取下稍冷却。用氨水中和至氢氧化铁沉淀完全并过量10 mL。加热微沸3~5 min,用定性快速滤纸过滤,用热的氨水洗液洗涤烧杯和沉淀各3~4次,用热水洗烧杯和沉淀各1次。
将沉淀用热盐酸(1+1)溶解于原烧杯中,用热水和热盐酸交替洗至滤纸为白色,溶液体积控制在100~125 mL,加入5 mL盐酸酸化后加热至烧杯底部有小气泡,趁热用氯化亚锡将溶液还原成淡黄色。冷却后,加入2~3滴钨酸钠溶液,用三氯化钛还原至“钨蓝”出现,用稀重铬酸钾(1 g/L)调节溶液至无色后(不计入体积)加入15~20 mL硫磷混酸,4~5滴二苯胺磺酸钠指示剂,用重铬酸钾滴定至溶液呈紫色为终点。
回渣做补正的实验方法:将过滤后的滤纸置于聚四氟乙烯的烧杯中,用少量水润湿后加入10 mL盐酸、15 mL硝酸、2 mL氢氟酸、2 mL高氯酸,盖上杯盖,置于电热板上加热至样品完全分解,继续加热蒸至近干后取下稍冷却,用水吹洗杯壁,加入10 mL盐酸和少许水,加热至溶液微沸,冷却后将溶液转移至100 mL容量瓶中定容摇匀。用电感耦合等离子体原子发射光谱(ICP-AES)法测定铁的含量。依据仪器的灵敏度和抗干扰性,选择259.940 nm为铁的最优谱线。仪器参数见表1。
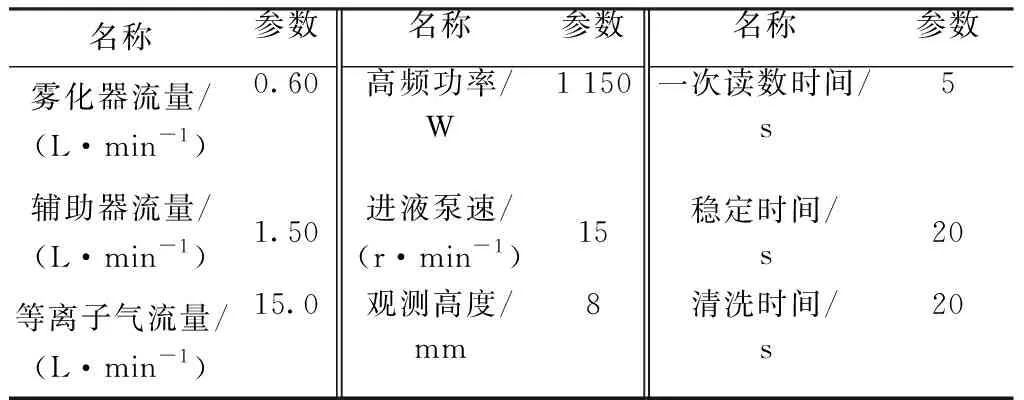
表1 仪器测量参数
1.3 空白实验
随同试料做空白实验,在用氯化亚锡溶液还原前,立刻用单刻度移液管加1.00 mL铁标准溶液,并按上述滴定步骤滴定,记下该滴定体积为V0,该滴定的空白值为V2=V0-1.00。
1.4 铁含量的计算公式
式中:
F——重铬酸钾标准滴定溶液对铁的滴定系数,mg/mL;
V1——滴定试液消耗的重铬酸钾标准滴定溶液的体积,mL;
V0——滴定空白溶液消耗的重铬酸钾标准滴定溶液的体积,mL;
m——称取试样量,g。
2 结果与讨论
2.1 溶样方法的选择
废杂铜中的元素主要有Cu、Pb、Zn、Fe、Al、Mn、Ni、As、Sb、Si、Au、Ag等[3]。采用不同溶样方法分别对1#、2#、3#、4#样品进行实验,考察样品溶解情况(表2)。

表2 溶样方法的选择
由表2可以看出,采用方法1溶解样品,四个样品均残余大量的不溶物。由于废杂铜样品基体复杂,本文采用称大样的方法解决样品不均匀问题,受称样量影响,方法1硝酸加入量过少,以铜为基体的试料不能充分溶解。方法2和方法3对2#和3#样品溶解不充分,有黑渣残余。过滤后回渣做补正,结果见表3。
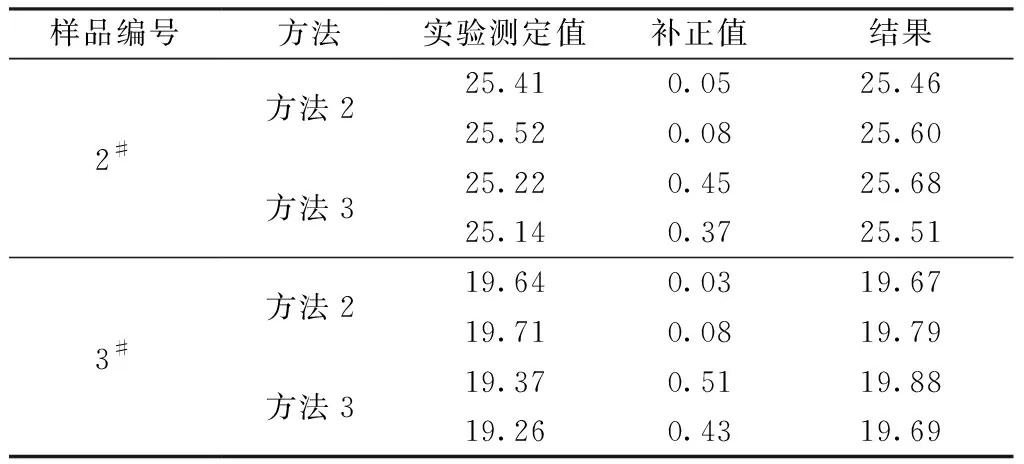
表3 不同方法实验结果比较
从2#和3#样品的结果来看,利用方法2溶解两个样品补正值均较低,说明样品溶解较为彻底,少量不溶黑渣中残留的铁含量很低。方法3的两个样品补正值均较高,说明样品溶解不彻底。方法2相比较方法3可以保证分析结果的准确性,因此采用方法2作为溶样方法。
2.2 氯化铵用量的选择
氨水沉淀分离法中通常加入NH4Cl等铵盐,增大溶液中的NH4+,使大量的NH4+作为抗衡离子,减少氢氧化物对其他离子的吸附,有效促进胶体沉淀的凝聚。准确移取10.00 mL铁标准溶液(铁含量为25 mg)5份,分别置于300 mL烧杯中,加入不同量的氯化铵,按实验方法进行测定,实验结果见表4。
结果表明,加入2~8 g的氯化铵,测定结果没有明显的差异,回收率均可得到满意的结果,综合考虑氯化铵的作用,实验选择氯化铵的用量为4~5 g。
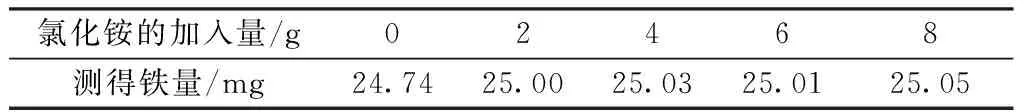
表4 氯化铵用量对测定铁的影响
2.3 氨水过量的体积
为使氢氧化铁沉淀完全,需要加入过量氨水,移取10 mL铁标准溶液(铁含量为25 mg)5份,分别置于300 mL烧杯中。按实验方法进行测定,加入氨水使氢氧化铁沉淀完全后,再分别加入不同量的过量氨水,结果见表5。
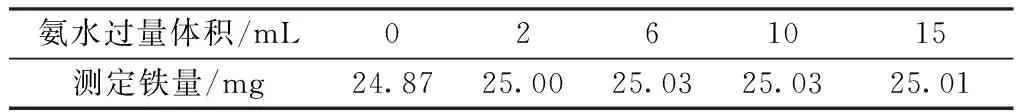
表5 氨水过量的体积
氨水过量时,不溶性物质转化成络离子溶解,从而与氢氧化铁沉淀分离,氨水过量2~15 mL时测定结果没有明显的差异,实验选择加入氨水过量10 mL。
2.4 硫磷混酸的加入量
滴定前加入硫磷混酸,一方面是保持溶液强酸性,以确保重铬酸根的强氧化性;另一方面利用磷酸根具有强的配位能力,加入H3PO4使终点更加敏锐。移取10 mL铁标准溶液(铁含量为25 mg)5份,分别置于300 mL烧杯中,按照实验方法进行测定,比较不同硫磷混酸的加入量对终点的影响,结果见表6。

表6 硫磷混酸的加入量
由结果可知,硫磷混酸的加入量影响终点的判断和终点溶液颜色。磷酸的加入与 Fe3+生成可溶性无色配离子,降低Fe3+浓度,使Fe3+/Fe2+的电位降低,化学计量点附近的突跃范围增大,终点误差减小,同时降低了Fe3+的黄色,更有利于终点的观察[7],提高测定结果的准确度。实验选择加入硫磷混酸15~20 mL可使终点敏锐,保证测定结果的准确。
2.5 精密度实验
选取4个废杂铜试样,按上述实验方法进行11次独立的测定,结果见表7,试样的相对标准偏差在0.38%~0.95%,该方法的精密度较好。
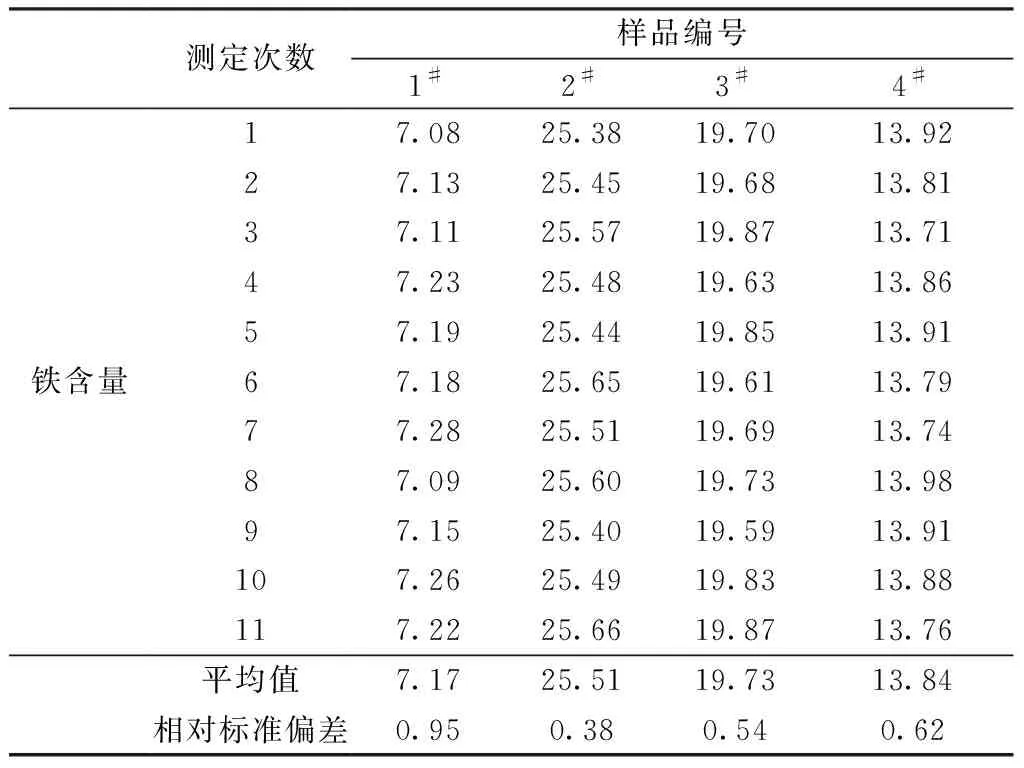
表7 精密度实验
2.6 加标回收实验
选取1#和4#两个废杂铜试样进行加标回收实验,按照实验方法进行铁的加标回收实验,结果见表8,由表8可知加标回收率在99.4%~100%,满足方法的准确度要求。

表8 加标回收实验
3 结论
通过实验表明,采用盐酸、硝酸、高氯酸溶解样品-重铬酸钾滴定法测定废杂铜中的铁含量,样品分解完全,氨水沉淀分离能够消除杂质元素的干扰。同时优化了前处理条件和滴定时硫磷混酸的用量。方法稳定、精密度高,分析结果准确可靠,能满足废杂铜中铁含量的测定需求。
