罕见47,XX,del(Y)(q11.2)/46,XX嵌合型克氏征1例报告并文献复习
2021-08-05汤冬冬叶四云贺小进张贤生
许 传 汤冬冬 耿 浩 叶四云 贺小进 张贤生
1.安徽医科大学第一附属医院妇产科生殖医学中心(安徽合肥 230088)
2.安徽医科大学生殖健康与遗传安徽省重点实验室(安徽合肥 230088)
3.安徽医科大学第一附属医院泌尿外科(安徽合肥 230088)
前 言
非梗阻性无精子症(non-obstructive azoospermia,NOA)一直是男性不育诊疗中的难题,约占无精子症患者的60%[1-3]。遗传学因素约占NOA病因的25%左右[4],其中染色体数目及结构的异常是最常见的遗传学因素。46,XX男性性反转综合征和克氏征是两种导致NOA的遗传学病因[4,5]。这两类患者临床表型相似,一般通过染色体核型分析进行鉴别。但由于47,XXY/46,XX嵌合型克氏征的存在,有时常规G显带法染色体核型分析难以鉴别这两类疾病。本文报告1例罕见47,XX,del(Y)(q11.2)[15]/46,XX[85]嵌合型克氏征导致的NOA病例,探讨其诊疗经验。
资料与方法
患者,男性,27岁,已婚,因“婚后未避孕2年未育”就诊于安徽医科大学第一附属医院生殖医学中心,3次独立的精液检查(禁欲3-7天,每次至少间隔1周)提示离心后均未检及精子;体格检查示:身高183cm,体重92.5Kg,第二性征发育不良,无喉结,无胡须,体毛稀少,双侧睾丸大小约2ml,阴茎发育不良;既往无隐睾及尿道下裂等病史。诊断:无精子症。完善相关检查:性激素检查,阴囊腹、盆腔超声探查,Y染色体微缺失检测及外周血染色体核型分析等。
结 果
一、患者性激素、阴囊腹、盆腔超声探查、Y染色体微缺失及染色体核型检查
血浆促卵泡生成素(FSH)44.78 mIU/ml,促黄体生成素(LH)22.19 mIU/ml明显高于正常范围,总睾酮(T)5.22 nmol/L低于正常值;阴囊超声提示双侧睾丸体积小(约2mL),双侧附睾及输精管未见异常回声,腹、盆腔超声未见明显异常(无卵巢、子宫及附件等组织);患者Y染色体微缺失检测提示:AZF6个经典位点(sY84、sY86、sY127、sY134、sY254、sY255)及SRY基因均存在;2次独立的外周血染色体核型检查(400带,G显带法,分析30个细胞)提示:46,XX。
二、患者父母染色体核型检查
在征得患者及家人同意后,对其父母行染色体核型检查,结果提示:父亲核型为46,X,del(Y)(q11.2)[21]/45,X[17]/46,X,Yqh-[2],母亲核型为46,XX,1qh+,见图1。

图1 患者父母染色体核型A-C:父亲核型46,X,del(Y)(q11.2)[21]/45,X[17]/46,X,Yqh-[2];D:母 亲 核 型46,XX,1qh+
三、患者染色体核型的再次鉴定
鉴于其父亲存在染色体嵌合、患者自身Y染色体AZF经典位点及SRY基因的存在,再次增加患者核型分析的细胞数目至100个,证实患者核型为47,XX,del(Y)(q11.2)[15]/46,XX[85],见图2,属罕见的嵌合型克氏征。
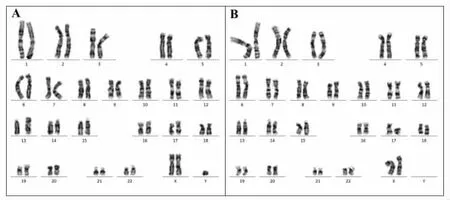
图2 患者染色体核型47,XX,del(Y)(q11.2)[15]/46,XX[85]
讨 论
一、46,XX男性性反转综合征
46,XX男性性反转综合征是一种罕见的遗传学疾病,该综合征最早由De la Chapelle[6]于1964年报道,故又称为De la Chapelle综合征。据统计新生儿发病率约为1/20,000[7]。其临床表现多样,按其表型可分为:男性表型、生殖器模糊表型和真两性畸形三大类[8]。另外,根据SRY基因是否存在,又可分为SRY基因阳性和SRY基因阴性两类。伴SRY基因阳性的男性性反转患者,其来源可能是Y染色体短臂上SRY基因在减数分裂过程中易位到X染色体上[9],该类患者往往表现为较明显的男性体征,但无胡须,无明显喉结,身高低于正常男性,外生殖器发育偏小,临床表型与克氏征相似。
二、Klinefelter综合征
克氏征是男性不育症中最常见的性染色体异常疾病[10],在新生男婴的发病率约为1/660[11],大约80%的克氏征患者表现为47,XXY核型,此外还存在其他类型的性染色体数目异常(48,XXXY;48,XXYY;49XXXXY)、染色体嵌合(如46,XY/47,XXY)及其他类型性染色体结构异常[12]。最常见的嵌合型克氏征患者通常携带两种细胞类型:46,XY/47,XXY。Paduch等人[13]的一项研究认为造成这一现象可能是由于胚胎在有丝分裂过程中X染色体未分离所致。
虽然SRY基因阳性的46,XX性反转与克氏征两类患者临床表型相似,但生育结局存在较大差异。46,XX性反转患者的生育选择仍局限于供精助育。而对于克氏征患者,随着显微外科技术在男科领域的应用,如今约50%的克氏征患者可以通过显微镜下睾丸切开取精技术找到精子,联合卵胞浆内单精子注射技术(ICSI)仍有希望生育属于自己的生物学子代[13]。因此,46,XX性反转综合征和克氏征的鉴别是十分必要的。
三、46,XX男性性反转综合征与克氏征鉴别诊断
临床上约90%的46,XX性反转患者携带有SRY基因[14],促使原始性腺向男性分化,表现为较明显的男性外生殖器,在青春期前无明显临床症状,故难以在早期发现并诊断。患者多数在成年后因不育、乳房发育异常或性腺功能减退而就诊[15],查体可见与克氏征患者相似的男性外生殖器特征,如双侧睾丸体积小,阴茎发育差,生化检查提示促性腺激素(FSH、LH)升高,睾酮水平降低。我们依据前2次外周血染色体核型分析、第二性征表现及激素检查结果,初步诊断该患者为46,XX男性性反转综合征。但该患者存在几点与46,XX性反转综合征不相符之处:(1)46,XX性反转患者多数携带有易位的SRY基因,但因无Y染色体存在,一般AZF检测表现为完全缺失;该患者AZF检测提示a、b、c区存在,考虑有Y染色体隐性嵌合的可能;(2)46,XX性反转患者多表现为身材矮小伴低体重,而该患者身材高大,体型偏胖,更偏向克氏征的表型。基于这两点考虑,我们与患者及家人沟通并征得同意,对其父母行染色体核型检查。结果提示:父亲核型为46,X,del(Y)(q11.2)[21]/45,X[17]/46,X,Yqh-[2]。在发现患者父亲存在染色体嵌合现象后,我们再次对患者行第3次外周血染色体核型分析并增加分析细胞数目至100个,最终证实患者为罕见的低比例嵌合型克氏征,核型:47,XX,del(Y)(q11.2)[15]/46,XX[85]。通常认为嵌合型克氏征的产生是由于部分细胞的X染色体不分离形成46,XY/47,XXY嵌合的现象。而此研究所报道的嵌合型克氏征的产生可能存在另一种机制。即父系配子(23,del(Y)(q11.2)或24,X,del(Y)(q11.2))与母系配子(24,XX或23,X)结合产生47,XX,del(Y)(q11.2)的合子。而这种异常合子在有丝分裂过程中部分细胞del(Y)(q11.2)丢失产生46,XX细胞,从而形成47,XX,del(Y)(q11.2)/46,XX嵌合现象。这一假设也在患者父亲身上得到了验证,其染色体核型也存在此类嵌合现象,即46,X,del(Y)(q11.2)[21]/45,X[17]/46,X,Yqh-[2]。查阅既往文献,这是国内首次报道家系中父子两代均出现有丝分裂过程中Y染色体异常丢失的复杂现象。其潜在机制有待进一步探讨,可能有助于为受累家庭提供新的、适用的遗传咨询方法。
四、常规染色体核型G显带法的不足与展望
染色体核型分析是遗传学疾病诊断的重要基础。常规的染色体G显带法是一种广泛使用的评价染色体异常的方法,通过Giemsa染色观察20-30个细胞分裂相的染色体核型[16]。但这种检查方法存在着一些不足,如染色体低比例嵌合、隐性易位、微缺失等不能及时发现,容易造成临床漏诊。通过增加核型分析的细胞数目可发现部分低比例嵌合的存在,这也是本研究中所应用的方法。此外,还可以通过荧光原位杂交技术、比较基因组杂交技术、微阵列-比较基因组杂交技术补充常规G显带法染色体核型检查方法的不足。
结 论
染色体核型异常是男性不育的重要病因,但临床常规使用的G显带分析法不能充分发现染色体低比例嵌合的存在,尤其是临床诊断中发现患者的染色体核型与表型不相符时,进行详细的家系分析结合Y染色体微缺失检查对于低比例嵌合型克氏征患者的诊断是十分必要。
