铝诱导法制备多晶硅薄膜的膜层间结合力影响机制
2021-08-03段永利臧浩天邓文宇齐丽君杜广煜谢元华刘坤
段永利,臧浩天,邓文宇,齐丽君,杜广煜,谢元华,刘坤
(1.沈阳中北通磁科技股份有限公司,沈阳 110168;2.东北大学 机械工程与自动化学院,沈阳 110819)
薄膜太阳能电池具有可靠性高、轻质、转化效率高等特点,特别适合大面积生产,具有广阔的市场前景[1-4]。多晶硅太阳能电池的间接制备方法主要包括金属诱导晶化法、激光诱导晶化法、固相晶化法及快速热退火法[5-8],其中铝诱导晶化法是目前生产常用的多晶硅制备方法之一[9]。在以往的研究中,研究人员多采用Al/α-Si 复合膜层结构作为该法制备的初始膜层,并通过低于Al/Si 共晶温度[10]的退火工艺进行处理,使复合膜层中α-Si 层的较高能量共价键Si─Si 键断裂,游离的Si 原子与Al 原子结合形成能量较低的Si─Al 键[11]。Si 原子不断地进入Al 层,当Al层中Si 原子的浓度达到饱和浓度时,将发生成核现象,即形成Si 晶核。Si 原子在Al 膜中的扩散持续供应Si 原子,以逐渐生长Si 颗粒(核),生长中的Si晶核取代了Al 原子,Si 原子与Al 原子的位置发生互换,直至多晶硅(poly-Si)膜生成才会结束,进而生成poly-Si/Al(Si)结构。目前,人们更多地是在Al 与α-Si 层中填入中间过渡层Al2O3,以起到提高多晶硅晶化效果的作用[12-13]。
关于铝诱导制备多晶硅的工艺已有大量的研究[14-17],但对复合膜层间结合力影响的相关研究比较匮乏。由铝诱导法的过程机理可知,退火时膜层间发生的置换反应会破坏膜层结构,这将十分考验所制备膜层的结合力。使用磁控溅射法镀覆Al/α-Si 层时,两层的结合力较好,但增加了中间过渡层后,少有人对Al2O3/α-Si 膜层间的结合力性能进行对比以及机理分析。王建军等[18]采用铝诱导法制备多晶硅薄膜,并通过氩离子轰击,提高了膜层结合力。邱春文[19]研究认为,在制备复合膜层过程中,辉光放电产生的等离子体对基体与膜层表面有清洗作用,从而可提高膜层结合力。本研究使用磁控溅射法进行了Al/α-Si 膜层结构与Al2O3/α-Si 结构的对比实验,并使用划痕仪测量两种复合薄膜的膜层间结合力。在膜层间结合力机理研究方面,本研究作为铝诱导制备多晶硅研究领域的先例,采用第一性原理计算观察膜层结合过程中的原子吸附点位、电子转移以及态密度变化等特性,计算出仿真过程中原子间作用力、原子位移量、原子间电子转移量以及吸附能等数值,从而分析膜层结合力影响机理,并阐述相应结论。
1 实验
1.1 基体预处理
基片选择康宁AG 玻璃,基片尺寸为10 mm×10 mm,厚度2.2 mm。用蒸馏水清洗基片5 min,然后在乙醇(99%)溶液中进行超声冲洗,用氮气干燥。将基板放置在样品架上,使用氩等离子体的附加溅射工艺施以5 min 溅射,以确保基板表面在沉积之前足够清洁[20]。
1.2 膜层制备
通过RF 磁控溅射机,在超高真空多室(旋转泵和涡轮分子泵组合)沉积系统(FJL560CI1)中,分别制备Al/α-Si 复合薄膜与Al2O3/α-Si 复合薄膜。其中Al 层使用直流磁控溅射法制备,Al2O3与α-Si 膜层采用射频磁控溅射法制备,各层制备工艺参数如表1所示。

表1 工艺参数设置Tab.1 Process parameter setting
1.3 样品表征
利用WS-92 型划痕仪获取两样品的膜层结合力,并进行对比分析。压头材料为金刚石,型号为Rockwell 型,最大弯曲半径为100 μm,锥度120°,线性应力加载范围为0~28 N,加载效率为9323.33 mN/min,最大划痕测量长度为3 mm。
2 结果与分析
根据所设置参数进行实验,制得的薄膜各层厚度均约为1 μm。对样品进行膜层结合力的测试,测试结果如图1 所示。由图1 可知,在Al/Si 样品的测量结果中,当划痕长度达到800 μm 左右时,产生了强烈的声信号,说明此时仪器指针划破最外层薄膜Al,并进入Si 层,此时指针施加载荷为7.80 N。
更多的样品测量结果如表2 所示,与图1 结果一致,在相同的制备工艺下,Al/Si 膜层临界载荷高于Al2O3/Si 膜层临界载荷。临界载荷的大小代表膜层结合力的大小[21],因此由测量结果可以看出,尽管中间缓冲层的加入提高了晶化率,并改善了晶化效果,但中间过渡层并没有对膜层结合力造成正面影响,甚至降低了膜层结合力。前人在研究中认为,几乎所有金属都能与Si 表面发生反应,生成硅化物[22],而硅化物的形成伴随着化学键的产生,因此增大了Si/Al 表面的结合力,这与本研究得出的结果一致。

图1 结合力测试结果Fig.1 Results of bonding force test

表2 Al/Si 与Al2O3/Si 样品膜层结合力对比Tab.2 Comparison of film adhesion between Al/Si and Al2O3/Si samples
影响膜层结合力的原因有很多,本研究忽略镀膜工艺、人为测量误差等影响因素,从原子间作用力的微观角度分析其原因。Si 与大多数金属单质的吸附过程中,都会存在化学键的形成,这说明了Si/Al 界面层可能存在着金属键的连接作用,这增大了Al 原子与Si 层表面的吸附能,提高了两膜层间的结合力。当Si 与Al2O3层接触时,O 元素的加入使得部分Si元素产生金属性,将会与Al 元素产生排斥力,因此二者不易吸附。第二种可能的原因是,两膜层之间并不产生化学键,二者没有确切的微观吸附点位,因此Si/Al2O3结合力不如Si/Al 膜层好。
为详细研究其过程机理,本研究采用第一性原理计算分析膜层微观行为。
3 第一性原理计算与分析
3.1 模型构建
为明确Si 原子分别在Al 表面和Al2O3表面的吸附情况,本研究运用密度泛函理论对相关体系的电子结构进行了观察和研究,并采用cambridge sequential total energy package(CASTEP)模块对相关的数据进行计算[22-23]。在运算过程中,主要以GGA 梯度上的PW91 泛函数值对不同电子之间的交换过程进行描述[24],并运用Mokhorst-Pack 法从不同的点位上进行取样[25],平面上停留的波长强度为300 eV。在布里渊区进行取样时,选取的K 点样本结构为5×5×1。在计算结构体系中的所有电子、原子与电子件的库伦吸引势能的过程中,主要以超软赝势法为主[26]。在开展自洽计算的过程中,得出的源自总能收敛系数为2×10−5eV/atom,在假设平均原子力小于0.5 eV/nm 的情况下,计算出的最大原子位移范围误差在0.02 nm以内。
首先分别对Al(001)晶胞及Al2O3(001)晶胞进行优化,模型建立如图 2 所示。二者超胞大小均为1×2×1,在表层上方预留1 nm 真空层,得出基底模型。其中紫色原子为铝原子,红色原子为氧原子。
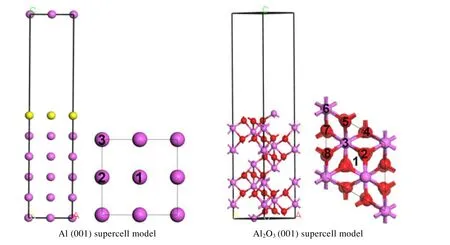
图2 超胞主/俯视图Fig.2 Supercell main/top view
3.2 吸附位置确认
进行优化之后得出的Al(001)与Al2O3(001)模型存在多个层次的原子结构,每个层次上的原子可保持运动状态。由于本实验中Si 在Al 以及Al2O3表面均为非晶结构,因此不考虑在不同材料表面制备的Si晶体结构对膜层结合力造成的影响。
Si 原子吸附在基底模型表面结晶的过程中,在Al(001)表面有3 种可能的吸附位点,在Al2O3(001)表面有8 种可能的吸附位点,具体位点已在图2中标注。
为明确Si 原子分别在两种表面是否具有更加稳定的吸附位,本研究对不同吸附位置上的吸附能进行了计算,计算结果如表3 及表4 所示。
吸附能源于原子在表面分离时需要耗费的能量。其中,Si 原子吸附于Al(001)表面的吸附能公式为:

式中,ESi/Al(001)为Si 原子吸附在Al(001)表面时产生的总能量;ESi表示Si 原子未吸附在Al(001)表面时的自有能量;EAl(001)表示集群状态下Al 原子所具备的能量。
通过观察吸附能的值,可以看出吸附效果的优劣。从表3 中的数据可知,Si 原子在Al(001)表面3个位置上产生的吸附能均为负值,表明该吸附结构具有较强的稳定性[27]。通过对三个吸附能大小的对比可以发现,Si 原子在2 位置上产生的吸附能绝对值大于其他位置,说明Si 原子在2 位置比其他位置更容易被吸附,因此认为Si 在Al(001)表面的最佳吸附位点为位点2。

表3 Si/Al 表面体系优化后的能量Tab.3 Energy after optimization of Si/Al surface system
对于Si 原子吸附于Al2O3(001)表面的情况,其吸附能公式为:

式中,ESi/Al2O3(001)为Si 原子吸附在Al2O3(001)表面产生的总能量;ESi表示Si 原子未吸附在Al2O3(001)表面时的自有能量;EAl2O3(001)表示集群状态下Al2O3分子所具备的能量。
由表4 可知,Si 原子在Al2O3(001)表面8 个位置的吸附能均为正数,说明Si 原子对Al2O3(001)表面具有排斥力,反映出吸附结构的稳定性较差,且吸附体系自身的稳定性也较差,Si 不会在Al2O3(001)表面吸附。

表4 Si/Al2O3 表面体系优化后的能量Tab.4 Energy after optimization of Si/Al2O3 surface system
为分析Si 原子不易吸附于Al2O3表面的原因,本研究通过电荷布局分析对电荷的转移情况进行定量讨论,分别对8 种吸附结构的Al、O 以及Si 原子的电子得失情况进行统计,情况如图3 所示。

图3 各类原子得失电子情况Fig.3 Electron gains and losses for various types of atoms
图3 中纵坐标为电子得失情况,正值表示失去电子,负值表示得到电子。根据图3 可以得出,在Si原子吸附于Al2O3表面的过程中,Al 原子失去电子表现为金属性,O 原子获得电子表现为非金属性。而对于Si 元素,我们已知其具有亲氧性,Si 遇O2易失去电子,呈现出金属性,但遇Al 时,易得电子呈现非金属性。因此在Al2O3晶胞中,不同点位下Si 原子得失电子情况不同,呈现的性质不同。其中,当Si 原子位于位点4、6、7 时,失去电子,呈现金属性;当Si 原子位于位点1、2、3、5、8 时,得到电子,呈现非金属性。
3.3 电子态密度
电子态密度图直接反映出不同原子与晶体表面的电子分布状态存在相互影响、相互作用效应。由前文已知,Si 原子在Al 层的最佳吸附位置为位点2。本研究对Si 原子在Al(001)表面的2 位置上的吸附强度进行了计算,得出了Si 原子在不同吸附状态下的PDOS 与TDOS(分波态与总波态密度),详情如图4。
图4a、b 分别为Si 原子吸附前后的分波态密度(PDOS)。由两图对比可以看出,Si 吸附Al 层前后,Si 价带中的3s 电子没有明显变化,但3p 层电子在−12~−7.5 eV 能量区间出现了明显的峰值,最终导致总态密度上升。这说明Si 吸附于Al 层的过程中,Si原子的3p 电子作用于该吸附过程。图4c、d 分别为Al 层原子吸附前后的分波态密度(PDOS)。由两图对比可知,吸附后Al 层原子3s 电子与3p 电子都发生改变。吸附前,Al 层电子的能量区间在−13~5 eV,吸附Si 原子后,Al 层原子能量区间缩小至−12~4.5 eV,而3s 和3p 电子的态密度值均有略微减少。图4e、f分别为吸附前后的Al 原子层和Si 原子的总波态密度(TDOS)。由两图对比来看,吸附前,电子的能量区间在−13~5 eV,吸附Si 原子后,Al 层原子能量区间缩小至−12~4.5 eV,而电子的态密度值均有略微缩小。综上,在Al 层吸附Si 原子的过程中,主要是Si原子的3p 轨道电子和Al 原子的3s、3p 轨道电子起到吸附作用,吸附后,Si 原子3p 轨道电子的电子态数量增多,说明该轨道中能量较低的电子在吸附后增大了能量。
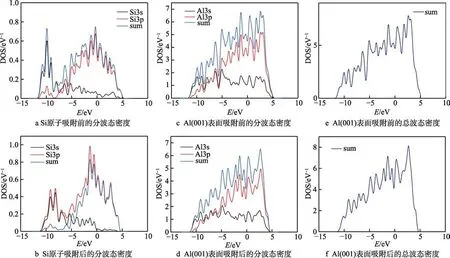
图4 Si 原子和Al(001)表面位置2 吸附前后的态密度(DOS)Fig.4 State density of SiO before and after Si atom and Al(001) surface position 2 adsorption: a) PDOS of Si atom before adsorption; b) PDOS of Si atom after adsorption; c) PDOS of Al(001) surface before adsorption; d) PDOS of Al(001) surface after adsorption; e) TDOS of Al(001) surface before adsorption; f) TDOS of Al(001) surface after adsorption
同时,为分析Si 原子吸附Al2O3表面的吸附效果影响,本研究得出了 Si 原子在不同吸附状态下的PDOS 与TDOS,详情如图5。
图5a、b 分别为Si 原子吸附前后的分波态密度(PDOS)。由两图对比可以看出,在Si 吸附Al2O3层之前,能量分别集中在−9~−6 eV、−3~−2 eV、−1~4.5 eV能量区间;吸附后,能量划分不明显,主要集中在费米能级处。这说明在吸附后,Si 原子更主要地呈现出金属性。图5c、d 分别为Al 层原子吸附前后的分波态密度(PDOS)。由两图对比可知,吸附后,Al2O3的3s 和3p 层电子能量均有所上升,约为3 eV,说明吸附后的Al2O3层原子中电子更不稳定。图5e、f 分别为吸附前后的Al2O3原子层和Si 原子的总波态密度(TDOS)。两图对比来看,吸附后,电子能量整体提升。综上,在Al2O3层吸附Si 原子的过程中,部分Si 呈现出金属性,而Al2O3中的Al 原子也呈现出金属性。金属离子间的作用力降低了Si 与O 原子形成共价键的可能,并且在Si 原子接近Al2O3后,Al2O3分子中电子能量攀升。这说明当Al2O3吸附Si原子时,Al2O3电子运动变得更不稳定,这与物理常识相违背,因此可认为Al2O3不吸附Si 原子。

图5 Si 原子和Al2O3(001)表面位置1 吸附前后的态密度(DOS)Fig.5 State density of Si before and after Si atom and Al2O3(001) surface position 1 adsorption: a) PDOS of Si atom before adsorption; b) PDOS of Si atom after adsorption; c) PDOS of Al2O3(001) surface before adsorption; d) PDOS of Al2O3(001)surface after adsorption; e) TDOS of Al2O3(001) surface before adsorption; f) TDOS of Al2O3(001) surface after adsorption
4 结论
1)当使用铝诱导晶化法制备多晶硅薄膜时,加入了中间过渡层Al2O3后,会降低膜层间结合力。
2)复合薄膜中Si/Al 界面层存在着Al─Si 金属键的连接作用。在Si 原子吸附Al 层的过程中,Si 原子的3p 轨道电子和Al 原子的3s、3p 轨道电子起到吸附作用,吸附后,Si 原子3p 轨道电子的电子态数量增多,说明吸附过程中产生了电子的转移,膜层间形成了硅化物。
3)Al/Si 复合膜层具有较强的稳定性,Si 原子在Al 界面层存在最佳吸附位点,Si 在Al(001)表面的最佳吸附位点为位点2。而Al2O3/Si 界面稳定性较差,Si原子在Al2O3表面受到排斥力,不存在最佳吸附点位。
4)在Al2O3层吸附Si 原子的过程中,部分Si原子呈现出金属性,而Al2O3中Al 原子也呈现出金属性,金属离子键的作用力降低了Si 与O 原子形成共价键的可能,因此Al2O3吸附Si 的能力低于Al。
