Identification of potential sex-related genes in Siniperca chuatsi*
2021-07-29QiaoyingZHUChongHANChengPENGXingniZHOUChongweiWANGLinqiangHANShuishengLIGuifengLIHaoranLINYongZHANG
Qiaoying ZHU , Chong HAN , Cheng PENG , Xingni ZHOU , Chongwei WANG ,Linqiang HAN , Shuisheng LI , Guifeng LI , Haoran LIN , Yong ZHANG ,**
1 State Key Laboratory of Biocontrol, Guangdong Provincial Key Laboratory for Aquatic Economic Animals and Southern Marine Science and Engineering Guangdong Laboratory(Zhuhai), School of Life Sciences, Sun Yat-Sen University, Guangzhou 510275, China
2 Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266373, China
3 Guangdong Key Laboratory of Animal Conservation and Resource Utilization, Guangdong Public Laboratory of Wild Animal Conservation and Utilization, Guangdong Institute of Applied Biological Resources, Guangzhou 510260, China
4 Guangdong Liangshi Aquatic Seed Industry Co., Ltd., Foshan 528100, China
Abstract Mandarin fish ( Siniperca chuatsi) is an economically important freshwater fish cultured in China. In this species, females grow faster than males. However, due to the lack of available genomic and transcriptome information, the mechanisms of sex diff erentiation remain poorly understood. In this study,Illumina high-throughput sequencing technology was used to sequence four cDNA libraries; the tissues examined included hypothalamus, pituitary gland, ovary, and testis. A total of 134 124 high-quality unigenes were obtained on average length of 1 361 bp and N50 of 3 312 bp. A search of all-unigene sequences against NR, NT, SwissProt, KOG, KEGG, GO, and InterPro databases resulted in 59 688 (44.50%), 76 329(56.91%), 50 432 (37.60%), 45 741 (34.10%), 48 760 (36.35%), 5 241 (3.91%), and 46 099 (34.37%)annotations, respectively. In a comparison of ovarian and testicular libraries, 15 289 ovary-biased genes and 10 035 testis-biased genes were identified, including a series of genes related to sex determination and diff erentiation, such as cyp19a1a, foxl2, sox9, dmrt1, amh, and others. In addition, 49 495 SSRs and 85 899 SNPs were detected in transcriptome data. Quantitative real-time PCR results of 15 sex-related functional genes indicated that RNA-seq data was reliable. This study will contribute to a better understanding of the molecular mechanisms of sex diff erentiation and development in Mandarin fish.
Keyword: transcriptome; Mandarin fish; HPG axis; female and male
1 INTRODUCTION
In almost all vertebrates, sexual reproduction is an important biological process, essential to maintain long-term survival and procreation. In order to ensure the continuation of species, vertebrates have evolved complex patterns of sex determination and diff erentiation. Fish are the most abundant vertebrates,distributed nearly in all aquatic environments, which contain more than 30 000 species (Froese and Pauly,2017). Fish have evolved all the strategies for sexual reproduction known in vertebrates, including parthenogenesis (Schartl et al., 1995), monoecism(Warner, 1984) and dioecism. The mechanisms of fish sex determination include two forms: genetic sex determination (GSD) and environmental sex determination (ESD) (Janzen, 1995). However, sex determination and diff erentiation remain poorly understood as they are extremely plastic and changeable, and even fish with GSD can be easily aff ected by environmental factors and exogenous hormones (Ospina-Álvarez and Piferrer, 2008).Currently, only a few master sex determination genes have been confirmed in several fish species:dmyin medaka (Oryziaslatipes) (Matsuda et al., 2002;Nanda et al., 2002),gsdfin medaka (Oryziasluzonensis) (Myosho et al., 2012),irf9yin rainbow trout (Oncorhynchusmykiss) (Yano et al., 2012),amhr2in fugu (Takifugurubripes) (Kamiya et al.,2012),dmrt1in half smooth tongue sole (Cynogossussemilaevis) (Chen et al., 2014) andamhyin Patagonian silverside (Odontestheshatcheri) (Hattori et al.,2012), and Nile tilapia (Oreochromisniloticus) (Li et al., 2015).
With the fast development of next-generation sequencing technologies, transcriptome sequencing is increasingly used to acquire gene expression data and to understand the associated mechanisms of regulation. Gonad transcriptome analysis has been used to identify genes related to sex diff erentiation and determination in many fish such as Nile tilapia(Oreochromisniloticus) (Tao et al., 2013), Japanese flounder (Paralichthysolivaceus) (Fan et al., 2014),Russian sturgeon (Acipensergueldenstaedtii)(Hagihara et al., 2014), spotted knifejaw (Oplegnathuspunctatus) (Du et al., 2017), fugu (Takifugutubripes)(Wang et al., 2017b) and channel catfish (Ictaluruspunctatus) (Sun et al., 2013). These data provided a lot of gonadal transcriptomic information and identified numerous sex-related genes, facilitating further studies on fish sex determination and gonadal diff erentiation. Moreover, previous studies have also demonstrated that the brain controls reproduction through the hypothalamic-pituitary-gonadal (HPG)axis and it influences gonad development (Weltzien et al., 2004; Sreenivasan et al., 2008). Thus, the hypothalamus and the pituitary gland are considered the tissues of choice to study the regulation mechanisms of sex determination and gonadal diff erentiation.
Mandarin fish, also known as Chinese perch(Sinipercachuasti), is one of the most important commercial fish, and has been widely cultured in China. It is in great demand from Chinese market because ofits high palatability and high content of essential amino acids and unsaturated fatty acids (Chu et al., 2010; Zhang et al., 2011). Mandarin fish is a demersal piscivore that feeds solely on live prey fish(Liang et al., 1998) and, although it grows fast, it is susceptible to diseases. For this reason, previous studies mainly focused on its growth (Wang et al.,2016; Tu et al., 2017), immunity (Wang et al., 2012,2014, 2017a) and prey preference (He et al., 2013,2018). In addition, female Mandarin fish grow faster than the males, therefore culturing all-female populations is a very eff ective approach to boost aquaculture production. However, only a few sexrelated genes have been cloned, such as the aromatase P450 gene (cyp19a) (Zou et al., 2017) and various growth hormone genes (Lu et al., 2008). In this study,the whole HPG axis transcriptome of Mandarin fish was sequenced using an Illumina HiSeq 2000 platform and numerous diff erentially expressed genes were identified in ovary and testis samples. This study was designed to provide a comprehensive understanding of the reproductive axis in Mandarin fish and to identify sex-related genes participating in sex determination and in gonadal diff erentiation. These data provide a valuable genomic resource and a large number of molecular markers to be used for further research on sex determination and gonadal diff erentiation in Mandarin fish.
2 MATERIAL AND METHOD
2.1 Sampling
Samples of the hypothalamus, pituitary gland,testis, and ovary were collected from freshly caught Mandarin fish (weight>500 g), frozen in liquid nitrogen and stored at -80 °C, to construct four independent libraries. The hypothalamus and pituitary gland samples of six Mandarin fish (3 males and 3 females) were mixed into a single sample. The testis and ovary samples from three diff erent individuals were also mixed into one sample.
2.2 RNA extraction and library construction
Total RNA was extracted from fresh frozen tissue using a TRIzol Reagent kit (Invitrogen, CA, USA)following the extraction protocol. Total RNA concentration, RIN value, 28S/18S and fragment size were measured using an Agilent 2100 Bioanalyzer(Agilent RNA 6000 Nano Kit), while purity was determined by ultraviolet spectrophotometer(NanoDropTM).
After total RNA was extracted, mRNA with polyA tail was enriched using magnetic beads with Oligo(dT), and an appropriate amount ofinterrupting reagent was added to the mRNA to fragment it at high temperature. One strand of cDNA was synthesized using the interrupted mRNA as template, and then double stranded cDNA was synthesized. After kit purification, recycling, and adhesive end repairing,base “A” was added to the 3′ end of cDNA and the joint was connected. Following fragment size selection, PCR amplification was performed. The constructed library was tested by an Agilent 2100 Bioanalyzer and ABI Step One Plus Real-Time PCR System, and by real-time PCR. Finally, the cDNA library was sequenced using Illumina HiSeq™. All procedures were performed in Beijing Genomics Institute, Shenzhen, China.
2.3 De-novo assembly and functional annotation
To ensure the reliability of the results, reads with joint contamination, unknown base N content greater than 5%, and low quality were removed before data analysis, and the filtered reads were named “clean reads” and stored in FASTQ format. Then, a de novo transcriptome assembly was carried out using Trinity software.
Functional annotation was performed through homology searches against the main public databases.All unigenes were compared with the sequences in the NCBI nucleotide (NT, ftp://ftp.ncbi.nlm.nih.gov/blast/db) database using BLASTn, the NCBI nonredundant (NR, ftp://ftp.ncbi.nlm.nih.gov/blast/db)protein database, the Clusters of eukaryotic Orthologous Groups (KOG, http://www.ncbi.nlm.nih.gov/KOG) database, the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg) and the SwissProt (http://ftp.ebi.ac.uk/pub/databases/swissprot) protein database using BLASTx or Diamond.
Blast2GO was used to identify the NR annotation results, and the number of unigenes related to each gene ontology (GO, http://geneontology.org) was calculated based on biological processes, cell composition and molecular function. InterProScan5 was used to annotate the InterPro (http://www.ebi.ac.uk/interpro) database.
2.4 Identification of diff erentially expressed genes(DEGs)
Based on the assembly results, clean reads of each sample were compared with each other using Bowtie2 software, and the gene expression level of each sample was calculated using RSEM. The diff erentially expressed genes (DEGs) between samples were detected using the PossionDis algorithm, which was defined as false discovery rate (FDR)≤0.001, and the gene with expression diff erence of more than 2 times.DEGs were classified and enriched for the GO function and KEGG pathway classification.

Table 1 Primers of qRT-PCR
2.5 Data validation by qRT-PCR
The reliability of transcriptome data was verified by qRT-PCR using LightCycler 480 SYBR GreenIMaster (Roche) and Light cycler 480 Real-Time PCR system (Roche). One denaturation cycle was performed at 95 °C for 10 min, and 40 amplification cycles were performed at 95 °C for 10 s,60 °C for 20 s and 72 ℃ for 20 s. The relative expressions of 15 DEGs in testis and ovaries,includingdmrt1,spata4,fshr,nanos2,sox9,cyp19a1a,foxl2,hsd17b1,hsd17b10,hsd17b12a,hsd17b8,spata2,spata5,star3, andstar5, were conformed using beta-actin as the reference gene. All samples were triplicated and double-tailed studentt-test was performed in confidence value of 95%(P≤0.05) to determine the significance of geneexpression. All primers were designed using Primer Premier 6 (Table 1).
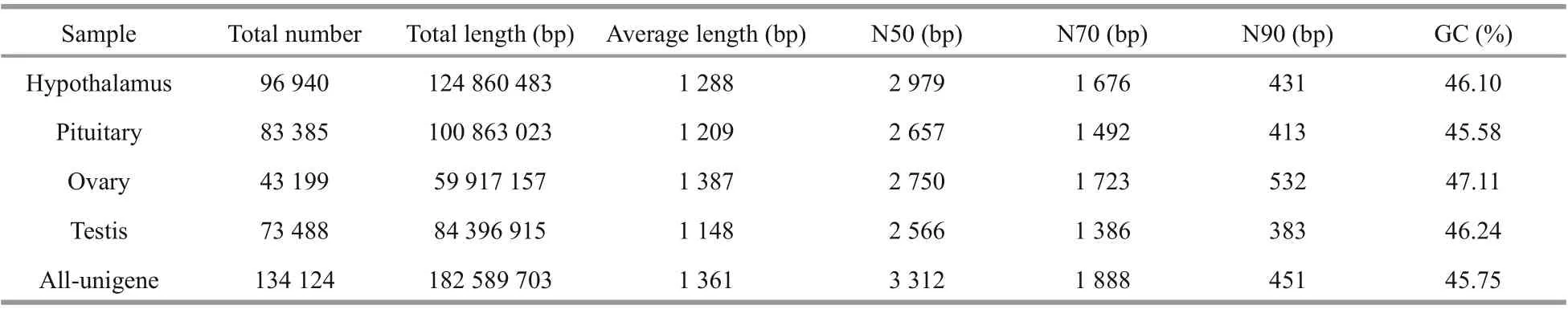
Table 2 Overview of the overall quality of the transcriptome
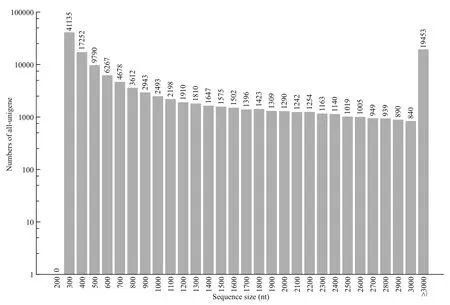
Fig.1 Length distribution of all-unigene
2.6 Identification of SSRs and SNPs
Simple sequence repeat (SSR) sequences were identified using MISAv1.0 (http://pgrc.ipkgatersleben.de/misa) in order to search for single nucleotide to hexanucleotide repeats. Parameters were set as follows: single base repeat at least 12 times, double base repeat 6 times, triple base repeat 5 times, quadruple base repeat 4 times, penta base repeat 3 times, hexa base repeat 2 times. When the distance between two microsatellites is less than 100 bp, a composite microsatellite is formed. HISAT(http://ccb.jhu.edu/software/hisat/index.shtml) was used to compare clean reads to unigenes in order to obtain single nucleotide polymorphism (SNP)sequences, and GATK (https://www.broadinstitute.org/gatk) was used to filter out low-quality SNPs with FS filter set at >30.0 and QD filter set at <2.0.
3 RESULT
3.1 Overview of transcriptome assembly quality
A total database of 27.56 Gb was analyzed using an Illumina HiSeq platform. Following the removal of assembly and redundancy, 134 124 unigenes were obtained, with total length, average length and GC content of 182 89 703 bp, 1 361 bp and 45.75%,respectively. The values of N50, N70, and N90 were 3 312 bp, 1 888 bp, and 451 bp respectively (Table 2).All unigenes were more than 300 bp in length, and 19 453 of them (14.50%) were more than 3 000 bp in length (Fig.1).

Table 3 Annotated gene distribution in seven public databases
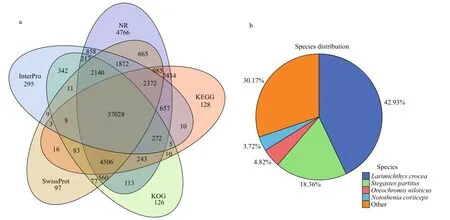
Fig.2 Venn diagrams depicting functional annotation and species distribution of homologous genes annotated in the NR database
3.2 Gene function annotation
Through sequence matching against seven public databases, an annotation analysis was conducted on the remaining 134 124 non-redundant unigenes, and 82 290 sequences were annotated. The percentage of annotated unigenes in the NT database (76 329;56.91%) was the highest, followed by the NR database (59 688; 44.50%), while the sequences annotated in the GO database were the lowest (5 241;3.91%) (Table 3). A total of 37 028 genes were annotated in five public databases: NR, KOG, KEGG,SwissProt, and Inter Pro (Fig.2a). The percentage of sequences consistent with all unigene BLASTx hits against other populations in the NR database showed thatLarimichthyscrocea(42.93%) has the largest amount of homologous sequences toSinipercachuatsi, followed byStegastespartitus(18.36%),Oreochromisniloticus(4.82%), andNototheniacoriiceps(3.72%) (Fig.2b).
To predict the functional classification of genes,possible pathways, and gene interactions, all unigenes were annotated in GO, KOG, and KEGG databases.
A total of 18 247 unigenes were assigned to three GO categories: biological processes, cellular component and molecular function. In the category of biological processes, cellular (2 764) and metabolic processes (2 234) were the most represented items. In the cellular component category, the most represented terms were cell (2 100) and cell part (2 080). In addition, in the molecular function category, the most abundant terms related to binding (2 543) and catalytic activity (1 866) (Fig.3a).
A total of 45 741 (34.10%) KOG-annotated genes were classified into 25 molecular families, with the most abundant distribution observed in “signal transduction mechanisms” (12 936 unigenes),followed by “general function prediction only”(11 266 unigenes); the smallest distribution was“coenzyme transport and metabolism”, with only 296 unigenes (Fig.3b).
A total of 45 979 (27.92%) unigenes annotated in KEGG were divided into six diff erent functional groups, among which the three exhibiting the largest distribution were “signal transduction” (9 131 unigenes), “global and overview maps” (4 820 unigenes)and “cancer: overview” (4 638 unigenes) (Fig.3c).
3.3 Simple sequence repeats (SSRs) and single nucleotide polymorphisms (SNPs)
A total of 49 495 SSR markers were identified from 31 707 sequences. Most were mono-nucleotide(12 482 unigenes), di-nucleotide (23 987 unigenes)and tri-nucleotide repeats (11 085 unigenes). The AC/GT motif was most abundant in dinucleotide sequences (17 494 unigenes), followed by the AG/CT motif (4 359 unigenes). The AGG/CCT motif was most abundant in trinucleotide sequences (3 317 unigenes), followed by the AGC/CTG motif (1 893 unigenes). In addition, there were 1 292 quadnucleotide, 372 penta-nucleotide, and 277 hexanucleotide sequences (Fig.4a).
In addition, a total of 85 899 SNPs were detected in transcriptome data. 24 426, 21 923, 14 414, and 25 136 SNP sites were detected from the four transcriptional libraries derived from hypothalamus,pituitary gland, ovary, and testis samples, respectively.The frequency of A-G transition was higher than C-T, and the frequency of A-T transversion was higher than other transversion frequencies (Fig.4b;Table 4).
3.4 Diff erentially expressed genes in ovary and testis samples

Fig.3 Gene function distribution annotated in GO (a), KOG (b), and KEGG (c) databases

Fig.3 Continued
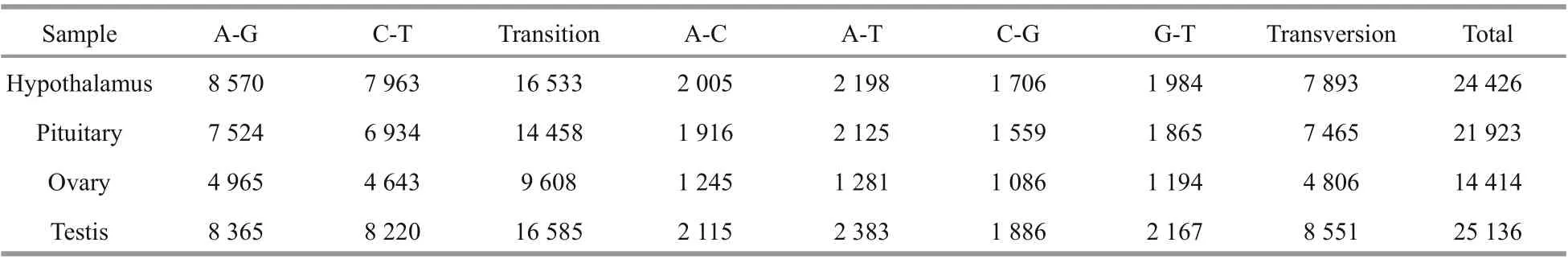
Table 4 Overview of SNP detection results
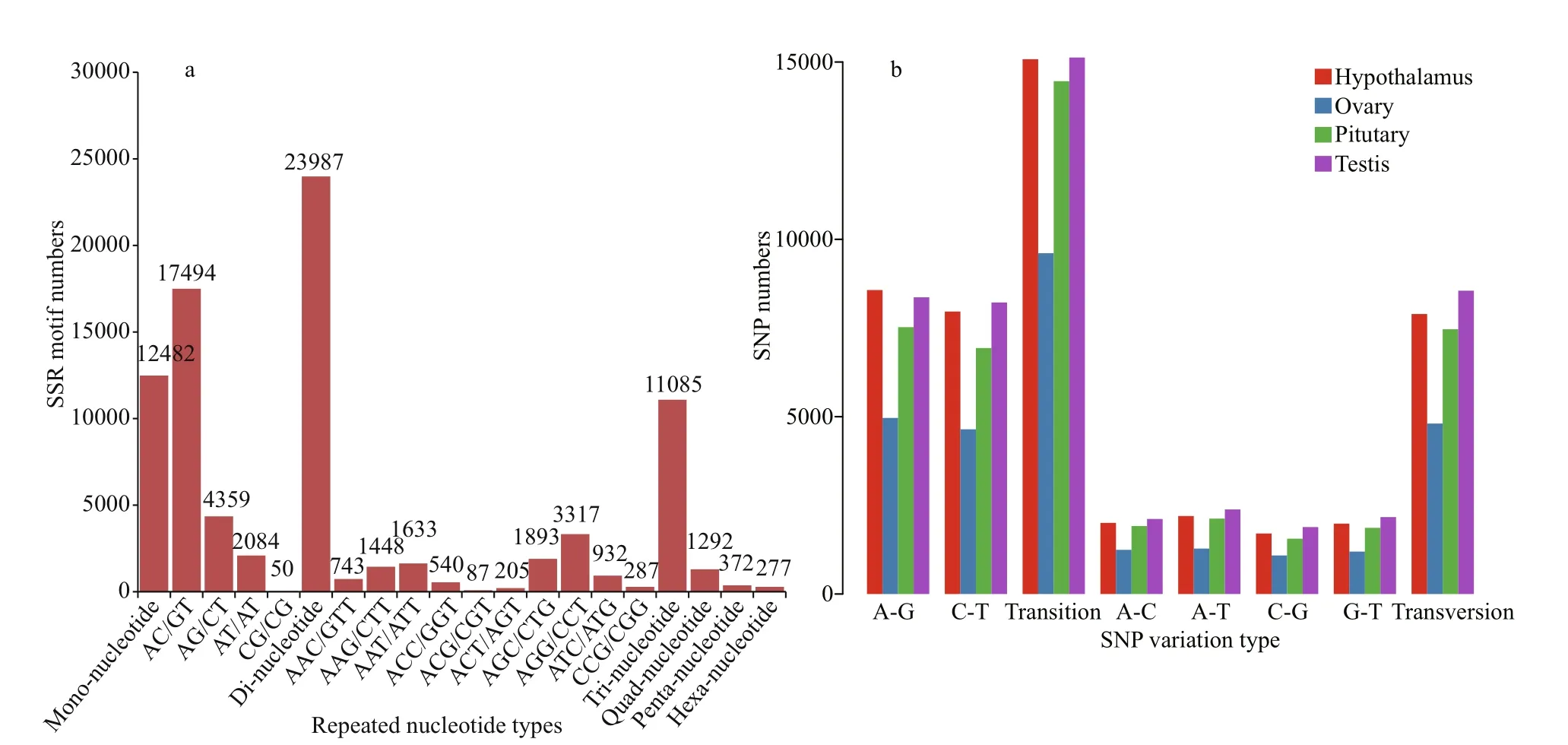
Fig.4 Distribution of SSRs and SNPs in the transcriptome of Siniperca chuatsi
A total of 25 324 DEGs were identified between the ovary and testis samples. Among them, 15 289(60.37%) were significantly higher in the ovaries(ovary-biased genes) and 10 035 (39.62%) in the testes (testis-biased genes). The remaining 47 035 genes were expressed in both testes and ovaries.Enrichment analysis of the GO signaling pathway showed that the annotation of DEGs to biological processes was predominant, followed by cellular components and molecular functions (Supplementary Fig.S1). In biological processes, 13, 13, and 3 unigenes were annotated to reproduction, reproduction processes, and hormone secretion, respectively(Supplementary Table S1). Enrichment analysis of the KEGG signaling pathway showed that DEGs were mainly distributed in metabolic pathways,endocytosis, and other processes (Supplementary Fig.S2). In addition, diff erent numbers of DEGs were annotated to the following pathways: 150 to GnRH,259 to Wnt, and 130 to TGF-beta. Among the pathways related to steroids, estrogen signaling,ovarian teroidogenesis, steroid hormone biosynthesis,and steroid biosynthesis were 189, 77, 50, and 27,respectively (Supplementary Table S2).
Based on the comparison of the functional annotation of Mandarin fish transcriptome sequences from various databases with the published data of other species, numerous genes related to the HPG axis and sex diff erentiation were identified(Supplementary Table S3), such as kisspeptins (kiss),gonadotropin-releasing hormone (gnrh),gonadotropins (gths), forkhead transcription factor2(foxl2), cytochrome P450 (cyp19a), doublesex and mab-3 related transcription factor 1 (dmrt1), SRY-box transcription factor 9 (sox9), anti-Mullerian hormone(amh).
3.5 Validation of transcriptomic data
Fifteen randomly selected DEGs between ovary and testis, includingdmrt1,spata4,fshr,nanos2,sox9,cyp19a1a,foxl2,hsd17b1,hsd17b10,hsd17b12a,hsd17b8,spata2,spata5,star3, andstar5,were used to verify the validation of transcriptomic data by qRT-PCR, and the results were consistent with the transcriptome data (Fig.5).
4 DISCUSSION
Mandarin fish, commonly known as Chinese perch,displays obvious sexual growth dimorphism that females grow faster than males from juvenile to commercial size. This means that all-female Mandarin fish cultures have greater economic benefits and broader prospects (Wang et al., 2006). However, due to the lack of genomic and transcriptome data and studies on sex-related genes, the molecular mechanisms of sex diff erentiation in this species are poorly understood. In this study, 134 124 unigenes were obtained by Illumina high-throughput transcriptome sequencing with N50, N70 and N90 values of 3 312, 1 888, and 451, respectively,indicating that the transcriptome data were of high quality and reliable. A large number of sex-related genes in the HPG axis of Mandarin fish were found,providing potential candidate genes for future research on reproduction, development and sexual diff erentiation.
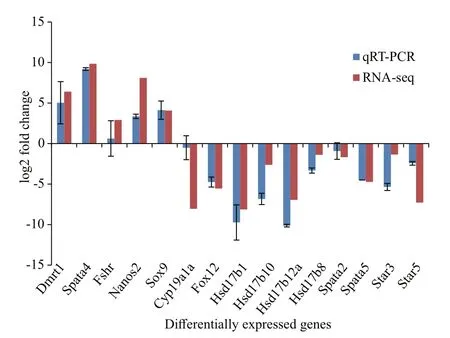
Fig.5 Validation of gonad transcriptome data by qRT-PCR
The HPG axis is the regulatory center of reproductive processes, and its signaling pathway is initiated by gonadotropin-releasing hormone (gnrh).This hormone displays dose-dependent induction by kisspeptin (kiss) (Novaira et al., 2009; Li et al.,2019b), and once it binds to the gnrh receptor (gnrhr),it stimulates the pituitary gland to secrete gonadotropins, including the follicle-stimulating hormone (fsh) and the luteinizing hormone (lh), to promote the production of sex hormones and the development of gametes. Due to the occurrence of fish-specific genome duplication (FSGD) events(Robinson-Rechavi and Laudet, 2001; Robinson-Rechavi et al., 2001), most fish species have two kinds of kisspeptin, and this is also true for Mandarin fish (Selvaraj et al., 2010). On the contrary, onlykiss2was found inSoleasenegalensis(Mechaly et al.,2011),TetraodonnigroviridisandGasterosteusaculeatus, suggesting thatkiss2may play a more important role. Previous studies confirmed that there are at least two or moregnrhs in teleost fish and thatgnrh3is unique to fish (Hildahl et al., 2011). As for gnrh receptors, the existence of two or moregnrhrs has been confirmed in teleost fish, and up to fivegnrhrs have been identified inFugurubripes(Moncaut et al., 2005) and inTetraodonnigroviridis(Ikemoto and Park, 2005). In the transcriptome of Mandarin fish, three types ofgnrhand three types ofgnrhrwere found. The selectivity of GnRH receptors to the GnRH ligand varies in diff erent species(Lethimonier et al., 2004), and the specific binding of the GnRH ligand to the receptor in Mandarin fish remains to be explored.
Gths can increase intracellular cAMP and promote the activation of the PKA subunit that regulates the expression of steroid hormone genes (Selstam et al.,1976; Reinhart et al., 1999; Stocco et al., 2005). It was suggested thatfshmay play an important role in thecyp19a1acontrol during ovarian diff erentiation through the synthesis of cAMP second messenger(Yamaguchi et al., 2007; Guiguen et al., 2010). The major sex hormones active in teleost fish are testosterone (T), 11-ketotestosterone (11-kt) and 17-estradiol (E2). Their synthesis starts with cholesterol. Cholesterol is transferred from the outer mitochondrial membrane to the inner membrane under the action of a steroidogenic acute regulatory protein (star) (Miller, 2007), and then is converted to androstenedione under the catalysis of P450scc,CYP17A1 and 3β-HSD. Subsequently, 17β-HSD3 catalyzes the conversion of androstenedione to T,which is further catalyzed to 11-Ketotestosterone by CYP11 and 11β-HSD2, and CYP19 catalyzes the formation of E2 from androstenedione T (Simpson et al., 1994). Our data showed that in the process of androgen synthesis, most of the genes coding for speed-limit enzymes were highly expressed in males,includingcyp11a1,cyp17a1,cyp11b, andhsd11β2. In contrast, during estrogen synthesis, CYP19 aromatase is the key rate-limiting enzyme, encoded bycyp19a1(includingcyp19a1aandcyp19a1b). In particular,cyp19a1aencodes gonadal aromatase, which is highly expressed in females and is mainly involved in estrogen production in Mandarin fish. The function of sex hormones is accomplished by binding to receptors.Several isoforms oferandarhave been found in Mandarin fish, includingera,erb1,erb2,ara, andarb, which are similar to isoforms observed in other fish (Ogino et al., 2018).
Since the activity of CYP19 aromatase directly determines the male to female hormone ratio,cyp19a1ais considered the central factor for sex diff erentiation in fish (Li et al., 2019a). Most of the sex-related genes found in fish are related tocyp19a1a.Foxl2is the earliest marker of ovarian determination and diff erentiation in vertebrates, and is highly expressed in female Mandarin fish. The transient transfection of fish showed thatfoxl2not only directly activated the transcription ofcyp19a1aand mediated the synthesis of estrogen (Yamaguchi et al., 2007),but it also bound to steroidogenic factor-1 (sf1/nr5a1)to enhance the transcriptional activity ofcyp19a1a(Wang et al., 2007). However, no sexual specific expression ofnr5a1aornr5a1bwas observed in Mandarin fish, whilenr5a2was highly expressed in the testis. Evidence proves the antagonistic eff ect ofnr5a1andnr5a2(Shi et al., 2019), but the role ofnr5a2in this process remains to be explored.
Dmrt1is a testis-biased gene that is highly conserved across animal phyla and was also highly expressed in male Mandarin fish. Previous studies have demonstrated thatdmrt1not only indirectly down-regulates the expression ofcyp19a1aby inhibitingfoxl2(Li et al., 2013; Lindeman et al.,2015), but it also directly binds to thecyp19a1apromoter to down-regulate aromatase activity (Wang et al., 2010).Dmrt1can promote the expression activity ofsox9(Wei et al., 2019), which has also an antagonistic eff ect onfoxl2(Nef and Vassalli, 2009;Georges et al., 2014). In Mandarin fish transcriptome data, only onesox9gene with high testicular expression was found, unlike most teleost fish that have bothsox9aandsox9b. In addition,amh, an important downstream gene related tosox9anddmrt1(Rodríguez-Marí et al., 2005; Webster et al., 2017),was shown to directly inhibit the expression ofcyp19a1a(Rouiller-Fabre et al., 1998), which is also highly expressed in the testes of Mandarin fish.
The combination of existing studies and transcriptome data from sex related genes with diff erential expression, suggests that Mandarin fish might share a similar regulation pattern for sex-related genes with other teleosts (Fig.6).
5 CONCLUSION

Fig.6 Regulation patterns of sex-related genes in Siniperca chuatsi
In this study, high-throughput RNA sequencing was used to provide transcriptome data ofSinipercachuatsi. The molecular mechanisms of gene function,reproductive regulation and gametogenesis in the HPG axis of Mandarin fish were predicted. A large number of candidate genes for gender preference have been identified, and these should be further studied to better understand their exact functions in these processes. In addition, a large number of SSR and SNP markers were found, providing basic information for the marker-assisted breeding ofSinipercachuatsi.
6 DATA AVAILABILITY STATEMENT
All raw reads of transcriptome sequencing data have been deposited at the NCBI Short Read Archive(SRA) database (SRA accession Nos.: SRR11743000,SRR11743001, SRR11743002, SRR11743003).
杂志排行

Journal of Oceanology and Limnology的其它文章
- Evaluating the eff ect ofinput variables on quantifying the spatial distribution of croaker Johnius belangerii in Haizhou Bay, China*
- Metabolomics analysis for skin ulceration syndrome of Apostichopus ja ponicus based on UPLC/Q-TOF MS*
- Four new species of free-living marine nematode from the sea areas of China*
- Virescentia guangxiensis (Batrachospermales, Rhodophyta):a new freshwater red algal species from South China*
- Molecular characterization and expression of the SiUCP2 gene in sea urchin Strongylocentrotus intermedius*
- Reproductive characteristics of Psilorhynchus homaloptera Hora and Mukerji, 1935 (Cyprinidae: Psilorhynchidae) in the lower Yarlung Zangbo River, Tibet*