GC-MS法测定地下水中28种半挥发性有机物
2021-07-29赵烔
赵 烔
吉林省地质科学研究所,吉林 长春 130012
0 引言
近年来随着我国工农业生产的快速发展,各种农药、除草剂、增塑剂等的大量使用,以及工农业废弃物的排放,产生大量的半挥发性有机污染物(Semi-volatile Organic Compounds,SVOCs)。SVOCs种类繁多,成分复杂,其中多环芳烃类、硝基苯类、有机氯、有机磷、酚类等是其重要的组成部分[1]。
目前,测定水中半挥发性有机物的前处理方法主要有固相萃取[2-4]和液液萃取[5-6]。固相萃取技术虽然能够简化前处理,节省试剂,但由于固相萃取小柱不同填料吸附能力不同,使其对不同性质的有机物具有选择性,测定不同种类的SVOCs时,很难使所测项目同时获得较好的结果。且市售固相萃取小柱因其厂家和批次不同,容易导致所测结果产生差异。液液萃取能够规避这些问题,可以同时萃取不同种类几十种半挥发性有机物,操作简单,适合多类别SVOCs广谱性检测。
我国2017年11月开始实施地下水地质矿产行业标准(DZ/T 0307-2017),标准检测项目中规定了27种半挥发性有机污染物的监测。其中包括了多环芳烃类、酚类、硝基苯类、有机氯、有机磷等。目前,我国还没有同时检测地下水中这27种半挥发性有机物(SVOCs)的国家标准方法。本文尝试建立一种广谱性的液液萃取-气相色谱质谱联用方法,通过内标法定量,同时检测水中28种SVOCs,覆盖了DZ/T 0307-2017所列除多氯联苯外全部的27个SVOCs检测项目,为相关测试工作提供技术参考。
1 实验部分
1.1 仪器和试剂
日本SHIMADZU GCMS-QP2020气相色谱质谱联用仪,配备AOC-20i自动进样器;色谱柱型号:DB-5MS UI毛细管柱(30 m×0.25 mm×0.25 μm);莱伯泰科氮吹仪;Milli-Q纯水系统(美国Millipore公司);微量注射器、分液漏斗等常规分析器具。
二氯甲烷、乙酸乙酯、正己烷均为农残级。氯化钠和无水硫酸钠均为优级纯(使用前均于400 ℃烘干6 h)。
1.2 标准物质
28种SVOCs标准物质(o2si,100 mg/L):萘、敌敌畏、2,4,6-三氯酚、2,4-二硝基甲苯、2,6-二硝基甲苯、α-BHC、六氯苯、乐果、莠去津 、β-BHC、五氯酚、δ-BHC、百菌清 、蒽、γ-BHC、甲基对硫磷、七氯、马拉硫磷 、毒死婢 、对硫磷、荧蒽、p,p′-DDE、p,p′-DDD、p,p′-DDT、o,p'-DDT、邻苯二甲酸二(2-乙基己基)酯、苯并[b]荧蒽、苯并[a]芘。

2种替代物(SS)标准物质(o2si,2 000 mg/L):2-氟联苯、对三联苯-d14。
1.3 仪器条件
1.3.1 GC条件
进样口温度260 ℃;不分流进样;流量控制方式:恒线速度;柱流速1.0 mL/min;进样量1.0 μL;程序升温:初始柱温50 ℃,保持4 min,以10 ℃/min升至290 ℃,保持7 min。
1.3.2 MS条件
扫描方式:SIM模式;EI源,离子源温度230 ℃;接口温度280 ℃;电子能量70 eV;溶剂延迟7 min。
1.4 实验方法
取1 L水样于2 L的分液漏斗中,加入约30 g氯化钠,摇动溶解后加入一定量替代物,使水中替代物质量浓度为0.5 μg/L。加入60 mL二氯甲烷,液液萃取10 min,静置10 min,转移有机相。分别在水样pH<2、pH≈7和pH>11条件下,重复上述萃取过程,合并有机相,用装有无水硫酸钠的球型漏斗过滤除水,萃取液经旋蒸和氮吹浓缩至1 mL,加入内标,使其测试质量浓度为0.5 mg/L,用GC-MS测试,内标法分析定量。
2 结果与讨论
2.1 萃取溶剂选择
二氯甲烷具有广谱的溶解能力,对SVOCs有良好的溶解性。沸点低(39.8 ℃),容易浓缩,且毒性较低。另外二氯甲烷因其立体电子效应而有较好的反应惰性,一般不容易与目标物发生化学反应。在与水进行液液萃取分层后,位于下层,可直接收集,方便前处理萃取过程。因其良好的性质被很多国标方法及US EPA 3510C等方法推荐使用[8]。因此本文选择二氯甲烷为半挥发性有机物的液液萃取溶剂。
2.2 内标物和替代物选择
根据保留时间和性质相似原则,参考相关标准,选用2-氟联苯和对三联苯-d14共2种化合物作为替代物,能够较好的表征目标物回收率水平。
2.3 仪器条件优化
2.3.1 GC-MS系统性能测试
每次分析运行开始前,用DFTPP检查GC-MS系统是否达到性能指标要求。性能测试要求仪器参数为:电子能量70 eV,质量范围35~450 amu,扫描时间为每个峰至少5次,每次扫描不超过1 s。得到背景校正的DFTPP质谱后,确认所有关键质量数是否达到表1的要求[9]。
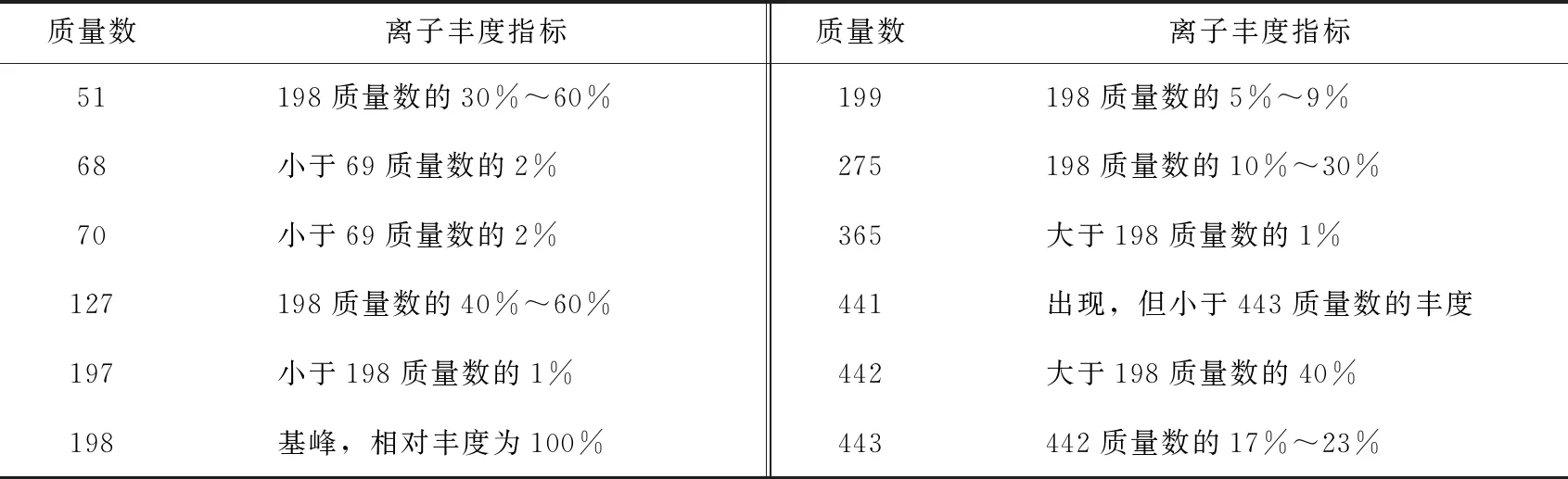
表1 DFTPP关键离子和离子丰度指标
测试时,每24 h进行一次连续校准,测定曲线中间浓度标准,连续校准测得浓度值与理论浓度值之间偏差小于20%时,可继续测试,否则需要检查仪器和标准品看是否需要重新维护和校准仪器或重新配制标准使用液。
2.3.2 GC-MS测试条件
28种目标物种类复杂,理化性质差异较大,虽然GC-MS定性定量不受保留时间相近峰的影响,但目标峰分离良好有利于总离子流程图的建立和后期数据分析处理过程。因此需要选择合适的色谱柱及合理设置气相柱温箱升温程序,使目标物能够在合理时间内全部出峰且分离良好。
色谱柱选择了通用的DB-5MS超高惰性柱,适用大部分半挥发性化合物和未知样品筛查。当气相柱温箱初始温度低于50 ℃时,运行样品时冷却时间会显著增加,且会使敌敌畏、2,4,6-三氯酚等目标物延迟出峰。因此为保证早流出峰的分离度,增加了初始柱温的保持时间。升温速率可以改变色谱图中间流出峰的分离度。升温速率高时,能减少样品运行时间,但中间组分如六氯苯、乐果、莠去津和β-六六六等不能完全分离。而升温速率低能提高分离度但将增加分析时间。结合初始温度、保持时间和升温速率,经过前期实验测试条件优化,确定GC条件为:初始柱温50 ℃,保持4 min,以10 ℃/min升至290 ℃,保持7 min,得到了很好的分离效果和峰形。
2.3.3 分离效果
在1.3的仪器条件下,直接进样1 μL标准混合溶液,采用SCAN模式获得34种SVOCs全扫描谱图,包括28种半挥发性有机物,2种替代物以及4种内标物,均得到较好的分离,无明显拖尾,色谱峰峰形较好,出峰时间理想。SVOCs总离子流谱图见图1。

图1 SVOCs标准样品的总离子流程谱图Fig.1 Total ion flow chart of SVOCs standard sample1.萘-d8(内标);2.萘;3.敌敌畏;4.2,4,6-三氯酚;5.2-氟联苯(替代物);6.2,6-二硝基甲苯;7.苊-d10(内标);8.2,4-二硝基甲苯;9.α-六六六;10.六氯苯;11.乐果;12.莠去津 ;13.β-六六六;14.五氯酚;15.γ-六六六;16.菲-d10(内标);17.百菌清;18.蒽;19.δ-六六六;20.甲基对硫磷;21.七氯;22.马拉硫磷;23.毒死婢;24.对硫磷;25.荧蒽;26.p,p′-DDE;27.对三联苯-d14(替代物);28.p,p′-DDD;29.o,p′-DDT;30.p,p′-DDT;31-d12(内标);32.邻苯二甲酸二(2-乙基己基)酯;33.苯并[b]荧蒽;34.苯并[a]芘
2.4 定性分析
根据目标化合物的特征离子、质谱图数据库,保留时间以及结合对目标物单标的定性分析,选择丰度高,质量数大的特征离子为母离子,选择高丰度的特征离子作为子离子。对34种目标化合物进行了定性分析。结果见表2。
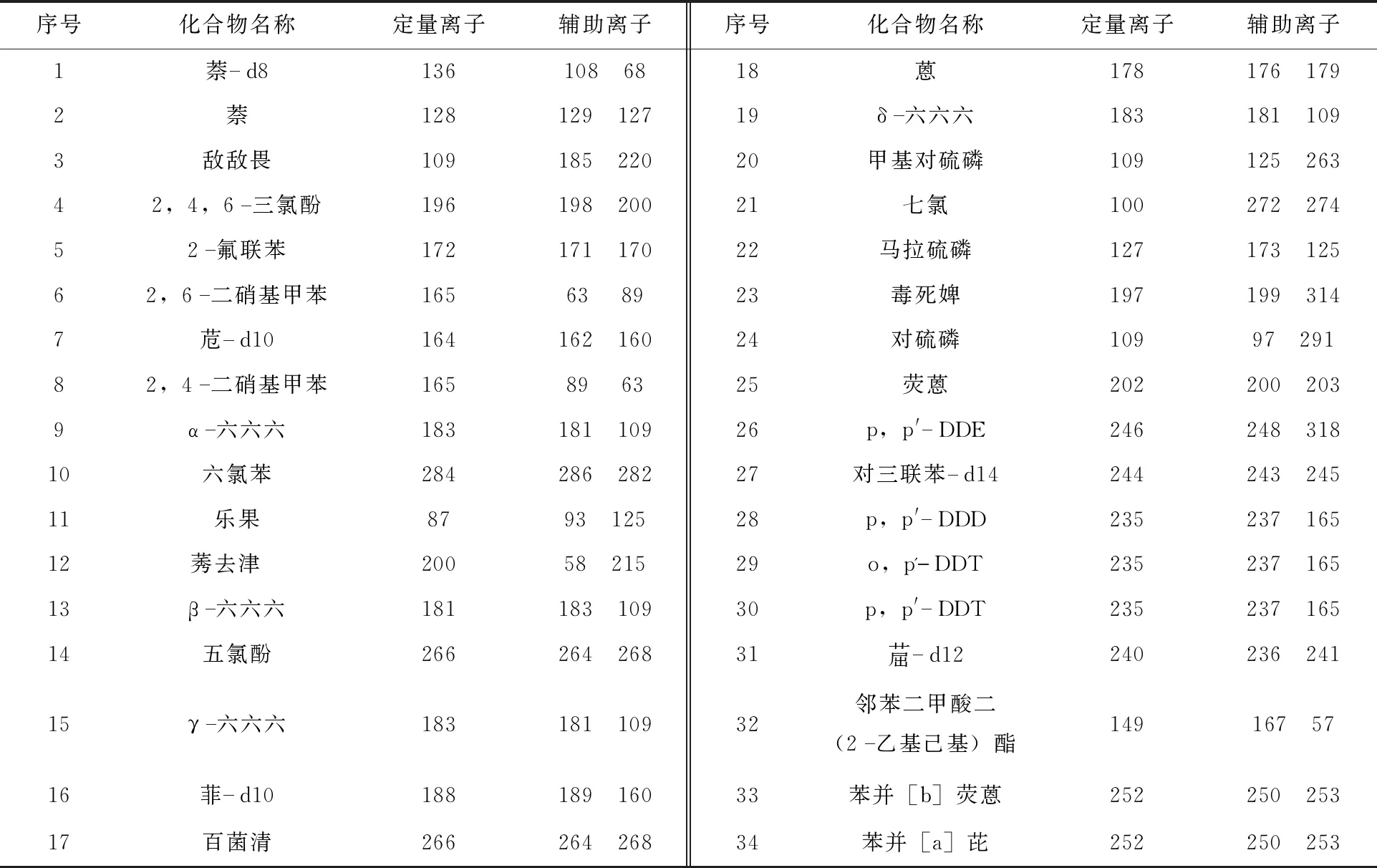
表2 目标化合物、内标和替代定性结果
2.5 定量分析
本文选用SIM模式进行定量分析,可以规避SVOCs测试过程中毛细管色谱柱固定相流失产生的柱流失峰、基线影响、非目标离子碎片对目标离子的干扰等问题,对表1中定量离子和特征离子进行选择性扫描,能够提高定量灵敏度。
将混合标准用二氯甲烷按比例稀释,配制成0.05, 0.1, 0.5, 1.0, 2.0 μg/mL 5个浓度点的标准系列,内标和替代物浓度均为0.5 μg/mL。采用内标法进行定量并拟合标准线性曲线。在SIM模式下,以标准溶液中目标化合物的峰面积与内标的峰面积比对目标化合物的浓度作图,得到目标物的定量校准曲线。实验条件下相关线性方程、相关系数、线性范围见表3。目标物在0.05~2.0 μg/mL线性范围良好,相关系数均大于0.999。
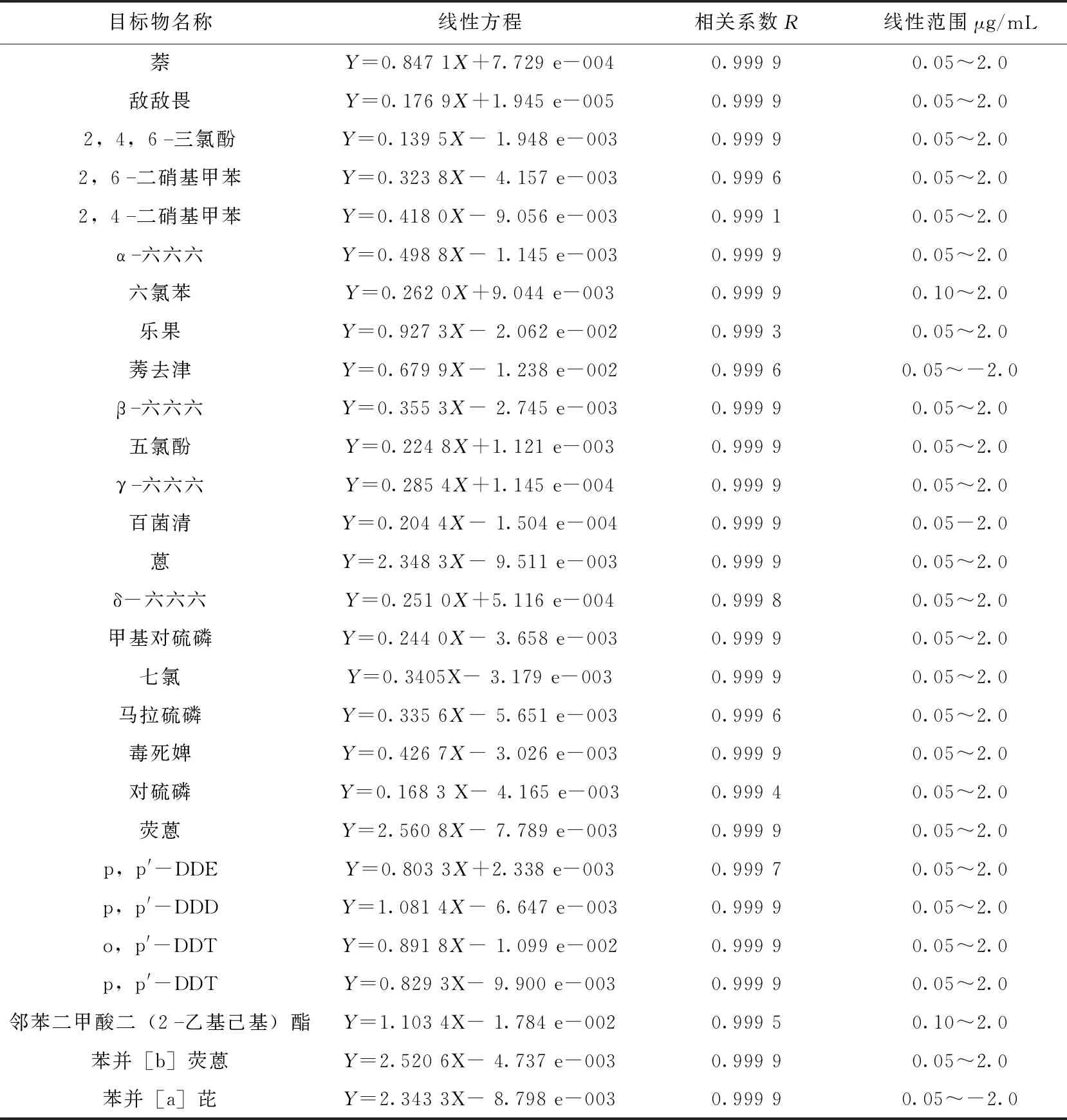
表3 28种SVOCs线性方程、相关系数和线性范围
待测样品溶液与标准溶液在相同分析条件下测定,根据样品溶液中目标物与内标物的峰面积比,由定量校准曲线得到该化合物的浓度。样品浓度(μg/L) =测定浓度(μg/mL)×萃取液体积(mL)/水样体积(L)。
2.6 检出限和测定下限
取1.0 L水样,加入SVOCs标准使用液,使目标物的加入量为0.05 μg,其中六氯苯与邻苯二甲酸二(2-乙基己基)酯为0.1 μg。按上述方法平行测定7份加标样品,计算7次测定结果的标准偏差S,方法检出限(MDL)和测定下限根据《HJ 168-2010》中的公式进行计算。检出限MDL=t(n-1,0.99)×s(n为样品的平行测定测试,s为7次平行测定结果的标准偏差),测定下限为4倍的检出限浓度。该方法检出限(MDL)和测定下限结果见表3。方法检出限范围为0.004~40.031 μg/L;测定下限范围为0.014~0.126 μg/L。低于DZ/T 0307-2017标准对不同目标物检出限要求。
2.7 准确度和精密度试验
在1.0 L水样中,分别加入线性范围内高低2个不同浓度的混标,使SVOCs加标量分别为0.1 μg和0.5 μg。每个样品重复测定7次,检验加标回收率及精密度,测定结果见表4。
目标化合物涵盖多环芳烃类、酚类、有机氯、有机磷以及硝基苯类等,不同物质允许的回收率范围不同,目标化合物在两个浓度点的加标回收率范围在68%~126%之间,目标物回收率较好,其中乐果回收效果一般,但符合标准要求,准确度满足测试要求。7个平行样的RSD在1.37%~9.20%之间,均低于10%,精密度符合要求。准确度精密度结果详见表4。

表4 28种SVOCs检出限和精密度结果
2.8 实际样品分析
分别选取了吉林省内送检的三个地区地下水样品(包括长春、四平和通化),进行了检测(样品编号为1,2,3),向1 L水样中加入0.5 μg替代物。用本文方法进行定量分析测试,样品质控措施完整,表5所示检测值为样品两次平行测定的平均值,三种水样中均未检出28种SVOCs,替代物和基体加标回收率均满足质控要求,两种替代物回收率控制图见图2。说明取样地区地下水样未受到半挥发性有机物污染。

表5 实际样品测试结果
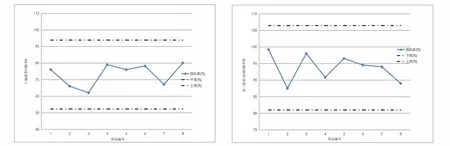
图2 两种替代物回收率控制图Fig.2 Control chart of recovery rate of two substitutes
3 结论
本文建立了一种广谱性液液萃取-气相色谱质谱联用法测定水中28种SVOCs的方法,方法投入低,简便,实用,在所选测试条件下, 28种SVOCs(涵盖DZ/T 0307-2017中的27种SVOCs)均可被良好分离和定量。
所测28种SVOCs在相应线性范围内,拟合标准曲线所得相关系数良好,R值均能超过0.999,符合实验测试要求。28种SVOCs的方法检出限范围为0.004~0.031 μg/L,测定下限范围为0.014~0.126 μg/L,远低于DZ/T 0307-2017的检出限要求,加标回收率范围为68%~126%,乐果回收效果一般,仍符合标准规定,准确度满足测试要求。RSD范围为1.37%~9.20%,均低于10%,精密度满足要求。
该方法具有灵敏度高、方法简便、投入少等优点,满足各项测试要求,为吉林省内地下水网中SVOCs的监测提供技术参考。
