赶黄草中大环多酚类成分含量测定及其提取工艺
2021-07-28江圣圭史鹏杰蔡舒心董志颖孙连娜空军杭州特勤疗养中心疗养三区药械科浙江杭州3000上海中医药大学中药学院上海003
陈 岚,江圣圭,史鹏杰,蔡舒心,董志颖,孙连娜 (. 空军杭州特勤疗养中心疗养三区药械科,浙江 杭州3000;. 上海中医药大学中药学院,上海 003)
赶黄草(Penthori Chinensis Herba)在民间又名水泽兰、水杨柳等,以其为原料制成的单味成方制剂肝苏颗粒现收载于《中华人民共和国卫生部部颁标准中药成方制剂十三册》附录[1]。赶黄草是苗族的传统药物,主产于四川古蔺,具有清热、利湿、解毒、活血、平肝、健脾等功效[2],可用于酒精性、非酒精性脂肪肝,淤积性、酒精性及其他诱导因素引起的肝损伤的保护和治疗[3-13]。赶黄草中化学成分类型众多,以黄酮类、萜类、酚酸类为其主要活性成分[14]。其中,具有以黄酮苷连接没食子酰基结构的大环多酚类化合物(图1)乔松素-7-O-[4'', 6''-(S)-六羟基二苯甲酰基]-β-D-葡萄糖苷(pinocembrin-7-O-[4'',6''-(S)-hexahydroxydiphenoyl]-β-D-glucose,PHG)、乔松素-7-O-[3''-O-没食子酰基-4'', 6''-(S)-六羟基二苯甲酰基]-β-D-葡萄糖苷(pinocembrin-7-O-[3''-O-galloyl-4",6''-(S)-hexahydroxydiphenoyl]-β-Dglucose,PGHG)、乔松素二氢查耳酮-7-O-[3''-O-没食子酰基-4'', 6''-(S)-六羟基二苯甲酰基]-β-D-葡萄糖苷(pinocembrin dihydrochalcone-7-O-[3''-O-galloyl-4'',6''-(S)-hexahydroxydiphenoyl]-β-D-glucose or thonningianin A,THA)的肝保护及抗肝纤维化活性较强[15-17],同时也具有较好的降糖活性[18],有较高的开发价值,故本实验建立HPLC 法同时测定该3 种化合物的含量,并通过正交试验优选其提取工艺,为该类成分的进一步开发研究提供前期基础。
1 仪器与试剂
1.1 药材来源
从四川收集3 批赶黄草药材(表1),药材经课题组孙连娜副教授鉴定为虎耳草科植物扯根菜(Penthorum chinensePursh)的干燥地上部分。各批次药材均留样于上海中医药大学中药资源与生物技术研究中心。
1.2 仪器与试剂
XS105DU 电子天平(瑞士Mettler Toledo 公司);XS104 电子天平(瑞士Mettler Toledo 公司);HDM-10000B 数显电热套(上海利闻科学仪器有限公司);N-1 300 旋转蒸发仪(东京理化器械株式会社);Milli-Q 纯水机(美国Millipore 公司);1 200 型高效液相色谱仪(美国 Agilent 公司);Centrifuge 5810R 高速台式冷冻离心机(德国Eppendorf 公司)。
PHG 对照品(批号:20 181 117)、PGHG 对照品(批号:20 181 103)、THA 对照品(批号:20 181 103)均由本实验室制备,且经HPLC 归一化法检测表明纯度均在98%以上;水为超纯水;甲酸(色谱纯,上海麦克林生化科技有限公司);乙腈(色谱纯,美国Thermo Fisher 公司);乙醇、甲醇(分析纯,上海泰坦科技股份有限公司 )。
2 方法与结果
2.1 大环多酚类成分的含量测定
2.1.1 对照品储备液的制备
精密称定PHG、PGHG、THA 对照品,加80%甲醇分别制成对照品储备液,质量浓度分别为0.610 4、0.604 4、0.485 2 mg/ml。
2.1.2 供试品溶液的制备
取赶黄草药材粉末(过3 号筛)1 g,精密称定后转移至250 ml 锥形瓶中,精密移取并加入80%甲醇水溶液100 ml,称重确定初始重量,回流提取1 h,放至常温,加溶剂补至初始重量,摇匀,取样,过膜,取续滤液作供试品溶液。
2.1.3 色谱条件
色谱柱:Agilent ZORBAX SB-C18柱(4.6 mm×250 mm,5 μm);流动相:乙腈(A)-0.5%甲酸水溶液(B);洗脱条件:梯度洗脱(0~20 min,32%→50%A,20~25 min,50%→90% A,25~30 min:90%→32% A);其他参数:流速为1 ml/min,柱温为30℃,检测波长为280 nm,进样量为10 μl。在此色谱条件下,供试品溶液(图2)中PHG、PGHG、THA 与其他成分均达到基线分离,分离度大于1.5,理论塔板数以PHG 计不低于20 000。
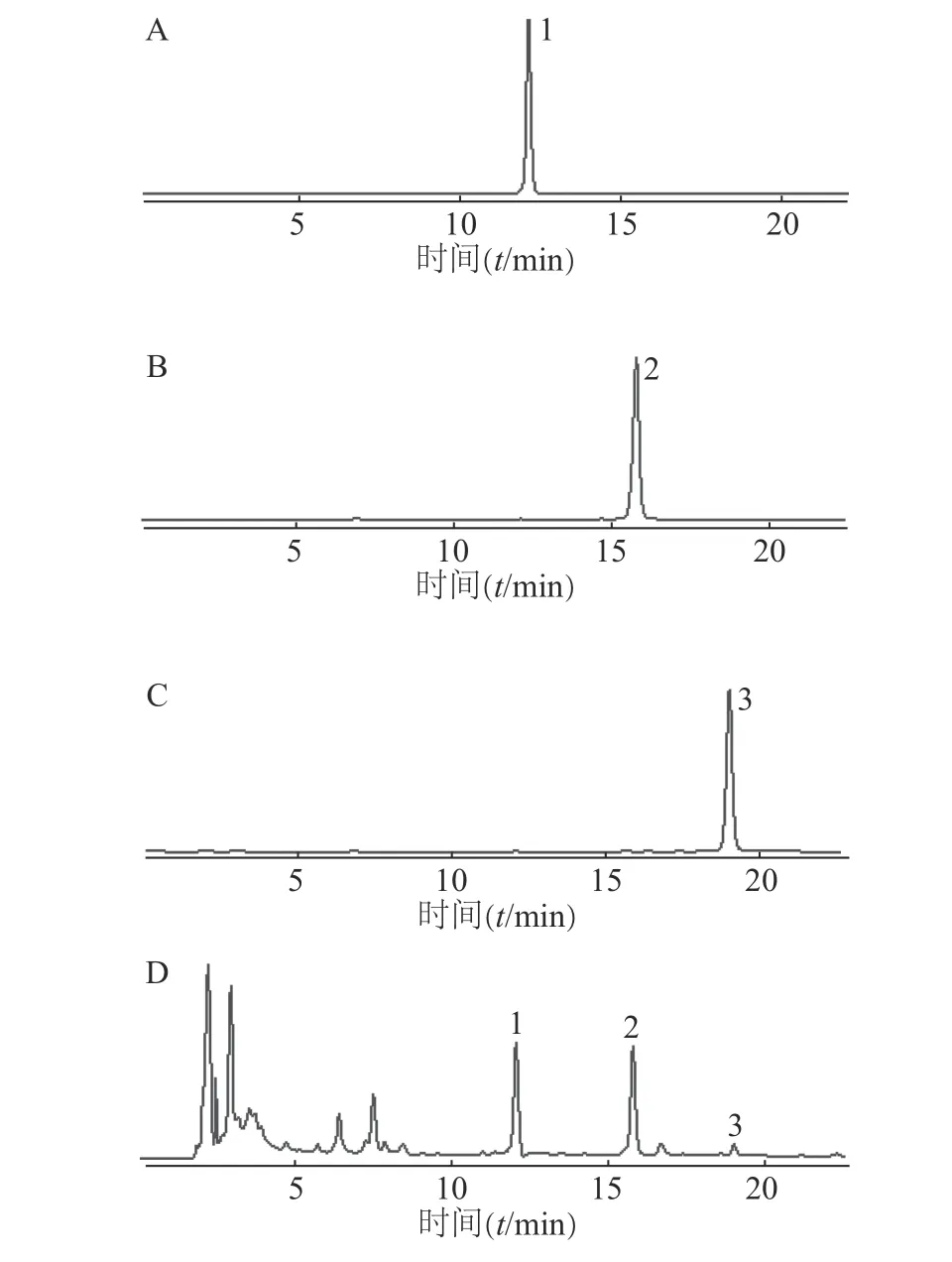
图2 标准品溶液(A-C)与药材提取液(D)HPLC 图
2.1.4 线性关系考察
取“2.1.1”项方法制备的混合对照品溶液,依次稀释2、4、10、50、100 倍,分别按“2.1.3”项色谱条件进样测定,以3 种成分的进样浓度(X)为横坐标,峰面积(Y)为纵坐标,分别进行HPLC 检测,线性拟合得回归方程。PHG、PGHG、THA 的回归方程分别为:Y=18 575.798X+5.091 9(r=0.999 9),Y=21 923.382X+29.293 3(r=0.999 9),Y=21 544.589X−13.093 6(r=0.999 9),线性范围依次是6.10~610.40、6.04~604.40、4.85~485.20 μg/ml,以上表明3 种化合物线性关系良好。
2.1.5 精密度考察
取对照品溶液,按“2.1.3”项色谱条件进行HPLC 检测,连续进样分析6 次,记录峰面积。结果显示PHG 峰面积的RSD 为1.15%,PGHG 峰面积的RSD 为0.18%,THA 峰面积的RSD 为0.12%,表明所用仪器精密度良好。
2.1.6 重复性考察
取赶黄草药材(S3,过3 号筛)6 份,按“2.1.2”项方法制备供试品溶液,再分别按“2.1.3”项色谱条件进行HPLC 检测,记录峰面积,计算含量。结果显示样品中PHG 的平均含量为5.83 mg/g,RSD为1.03%;PGHG 平均含量为9.99 mg/g,RSD 为0.91%;THA 平均含量为1.31 mg/g,RSD 为0.50%,表明本方法重复性良好。
2.1.7 稳定性考察
取赶黄草药材(S3,过3 号筛)1 份,按“2.1.2”项方法制备供试品溶液,分别放置0、4、8、12、24 h后取样,按“2.1.3”项色谱条件进样测定,记录峰面积。结果显示样品中PHG、PGHG、THA 峰面积的RSD 分别为1.72%、2.44%和4.06%,表明供试品溶液在24 h 内稳定。
2.1.8 加样回收率考察
取同一批赶黄草药材(S3,过3 号筛)6 份,每份约0.5 g,精密称定,按照药材含有量1∶1 的比例,分别精密加入PHG、PGHG、THA 对照品,按“2.1.2”项方法制备供试品溶液,再按“2.1.3”项条件进行HPLC 分析,记录峰面积,计算回收率。结果显示PHG、PGHG、THA 加样回收率分别为102.04%、100.90%、101.55%,对应的RSD 值分别为0.88%、0.82%、1.43%,表明该方法可靠。
2.1.9 赶黄草药材的含量测定
取各批次赶黄草药材(S1~S3,过3 号筛)各3 份,按“2.1.2”项方法制备供试品溶液后按“2.1.3”项条件进行HPLC 分析,记录峰面积并计算含量(表2)。
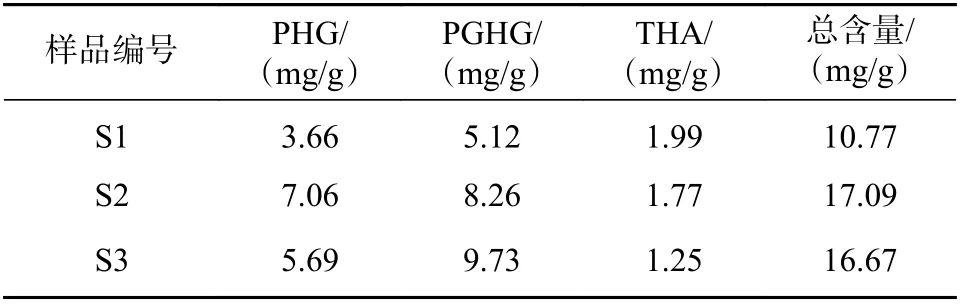
表2 赶黄草中3 种化合物的含量(n=3)
2.2 提取工艺的优化
2.2.1 提取方法考察
取赶黄草药材(S2)切成3~5 cm 小段,称取50 g,加入60%乙醇溶液500 ml,分别采用浸渍法(浸渍24 h)、渗漉法(浸泡24 h 后以5 ml/min 的流速收集渗漉液)、回流法(加热回流1 h)提取。结果表明,浸渍法平均总提取率为36.67%,渗漉法平均总提取率为36.82%,回流法平均总提取率为71.99%。考察结果为回流法提取效果最佳,因此选择回流法作为提取方法。
2.2.2 正交试验[19-21]
在确定提取方法为回流法的基础上,取赶黄草药材(S2),选择常规回流提取中对提取效果影响较大的因素:溶媒浓度(A)、提取时间(B)、溶媒用量(C)、提取次数(D)作为影响因素,每个因素各取3 个水平,在平行操作条件下,设计L9(34)正交试验(表3)。
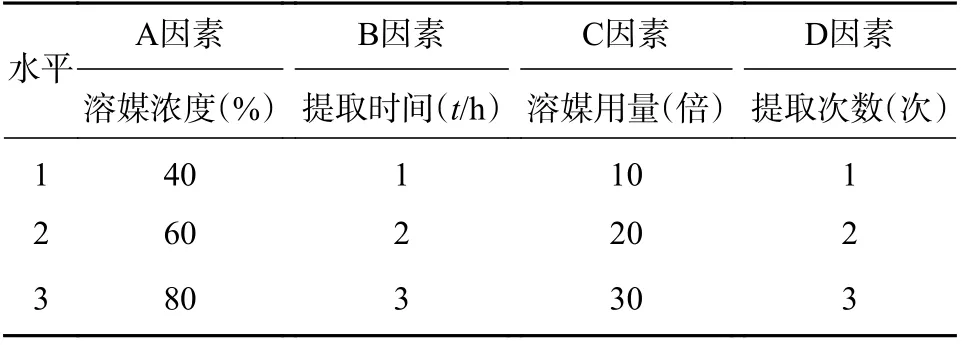
表3 正交试验因素水平表
以PHG、PGHG、THA 的总提取率作为考察指标得正交试验结果(表4),根据极差大小可以看出各因素对赶黄草中大环多酚类成分的影响大小为A>B>D>C;进一步方差分析(表5)结果表明,溶媒浓度(A)对提取效果有显著影响(P<0.05),而其他3 个因素无显著影响(P>0.05)。因此,根据正交试验得出的最佳组合为A3B1C3D2。
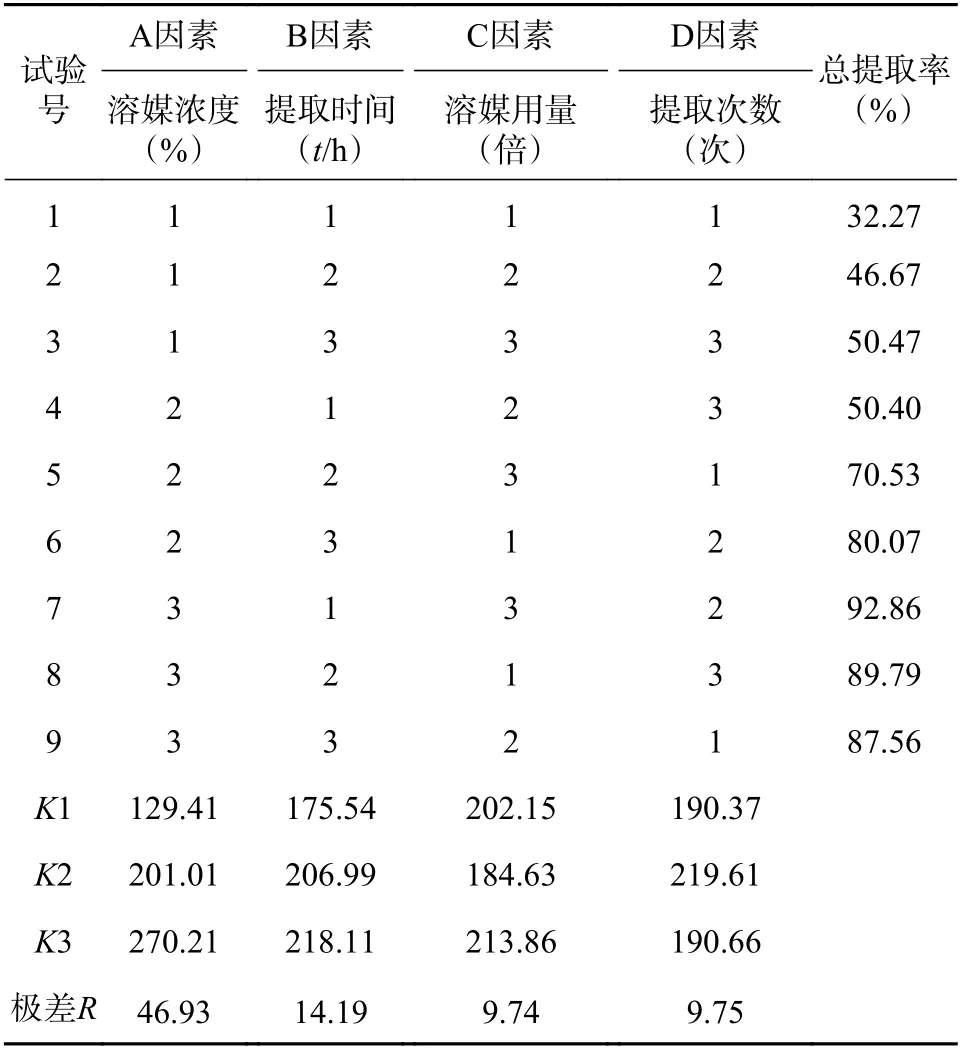
表4 正交试验结果表
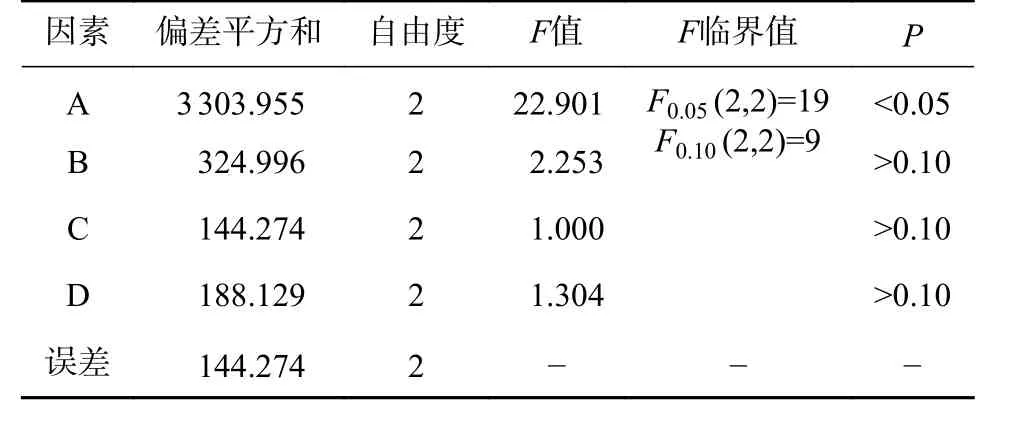
表 5 方差分析表
3 讨论与结论
本实验比较并优化了供试品溶液制备方法,并考察了不同型号色谱柱、不同检测波长、不同流动相组成,在此基础上建立了赶黄草中PHG、PGHG、THA 的HPLC 检测方法,该法能使样品中测定成分与其他成分达到有效分离,峰型好,可简便快速分析样品,检测结果准确可靠。
在建立含量测定方法的基础上进行提取工艺优化。从毒性大小、常用性角度选择乙醇为溶媒,单因素实验结果显示回流法提取效果最佳,因此选择乙醇热回流法作为提取方法,对常规热回流法影响较大的4 个因素进行3 个水平设置,建立L9(34)正交试验组,以便考察不同参数设定值对赶黄草中大环多酚成分提取率的影响。通过极差及方差分析发现溶媒浓度(A)即乙醇浓度对提取率有显著性影响,而其他3 个因素(B、C、D)均无显著性影响。经正交试验优选后的最佳提取工艺为A3B1C3D2,结合生产实际,从节约原料降低成本的角度,对无明显影响的3 个因素进行适当调整,调整后的提取工艺为A3B2C1D2,即取赶黄草干药材,切3~5cm小段,加入10 倍体积、浓度为80%的乙醇溶液,回流2 次,每次2 h。按照该工艺,取3 批药材各5 kg,进行3 次放大验证试验,提取率均在90%以上,且RSD 值为3.73%,说明该工艺稳定可行,可为赶黄草中该类成分的进一步开发研究打下基础。
