离子色谱-脉冲安培法检测食品中功能性低聚异麦芽糖
2021-07-23徐佳佳刘玉峰孔凡华乔子纯崔亚娟徐锡媛郑晓玮
徐佳佳,刘玉峰,孔凡华,乔子纯,崔亚娟,徐锡媛,郑晓玮,李 东
(北京市营养源研究所,北京 100069)
低聚异麦芽糖(Isomaltooligosaccharide,简称IMO)以淀粉或淀粉质原料,经酶法转化、精制、浓缩等工艺制得的一种淀粉糖产品,分子中至少含有一个葡萄糖分子间以α-1,6 糖苷键结合的异麦芽糖、潘糖、异麦芽三糖,以及其他四糖以上的低聚糖的总称,是近些年发展速度较快的一种功能性低聚糖[1−6]。商品低聚异麦芽糖产品规格主要有两种,IMO-50 型和IMO-90 型[7]。低聚异麦芽糖属于非消化性低聚糖类,难以被胃酶消化,具有水溶性膳食纤维功能,其热量低,基本上不增加血糖血脂,有助于维持肠道菌群平衡,改善腹泻与便泌,预防龋齿,提高机体免疫力[8−10]。鉴于低聚异麦芽糖的特性和保健功能,其已被日益广泛应用于保健品、食品、医药、化妆品等领域[11]。食品中加入低聚异麦芽糖对促进人体健康具有重要意义,一定程度上降低肥胖、糖尿病、冠心病等症的发病率。
糖类的检测方法主要有比色法[12]、毛细管电泳法[13]、色谱法[4,14−28]、滴定法[16]等。其中色谱技术是目前所广泛应用的检测糖的方法。目前,低聚异麦芽糖的测定方法主要有高效液相色谱法[4,14−17]、气相色谱法[18−19]、离子色谱法[20−28]等。高效液相色谱法受示差检测器的影响,灵敏度较低;气相色谱分离效果好,灵敏度高,但样品前处理步骤复杂,一般需要对目标物进行硅烷化反应,离子色谱法测定糖具有样品前处理简单,无需衍生,且专一性强、灵敏度高的特点,其用于测定糖类逐渐被关注[29−32]。糖能够在pH≥12 的碱性环境中解离成阴离子,在电极上发生氧化还原反应,用阴离子交换柱进行分离后,可以用脉冲安培检测器检测[10]。本研究样品经水溶解提取,酸沉淀蛋白后,采用离子色谱-脉冲安培检测器测定低聚异麦芽糖,以期为监管政策的落实和实施提供必要的技术支持和有力保障。
1 材料与方法
1.1 材料与仪器
异麦芽糖对照品(纯度97.0%)东京化成工业株式会社;异麦芽三糖对照品(纯度97.0%)东京化成工业株式会社;潘糖对照品(纯度99.0%)、50%氢氧化钠溶液(色谱纯)Sigma公司;磺基水杨酸(优级纯)天津市光复精细化工研究所;甲醇(色谱纯)
Fisher公司;ON GUARD ⅡRP柱(3cc)美国ThermoFisher公司;0.45 μm水系滤膜 天津津滕公司;实验室用水 Aquaplore 2C超纯水;实验所需样品 购于北京各大超市。
ICS-3000 离子色谱仪 美国ThermoFisher公司,配脉冲安培检测器金工作电极,Ag/AgCl参比电极;KQ-500DE型超声波清洗器 昆山市超声仪器有限公司;ME235s电子天平 精度0.1 mg,德国赛多利斯公司。
1.2 实验方法
1.2.1 溶液配制 9 种糖混合标准储备溶液的配制:分别准确称取50 mg(精确至0.1 mg)的异麦芽糖、异麦芽三糖、潘糖、半乳糖、葡萄糖、果糖、乳糖、蔗糖、麦芽糖对照品到50 mL容量瓶中,用超纯水配制成浓度约为1000 mg/L标准储备液,放置4 ℃密封可保存1 个月。
9 种糖混合标准工作溶液的配制:准确吸取9 种糖混合标准储备液1.0 mL于25 mL容量瓶中,用水定容至刻度,为9 种糖标准使用溶液。分别准确吸取9 种糖使用溶液1、2、3、5、10、20 mL于50 mL容量瓶中,用水定容。得到标准曲线工作液浓度分别为0.8、1.6、2.4、4.0、8.0、16.0 mg/L,用时现配。
冰醋酸水溶液(0.05 mol/L):准确吸取600 μL冰醋酸于水中,用水定容至50 mL,混匀。
1.2.2 样品处理
1.2.2.1 采样和试样制备 固体样品:抽取有代表性的样品,用四分法缩减取200 g,粉碎后过筛,混匀,装入自封袋中,备用。
液体样品:混匀,备用。
1.2.2.2 提取 称取0.5~5.0 g(精确至0.001 g)的制备好的样品于100 mL容量瓶中,加入80 ℃左右的热水40 mL,超声提取15 min,冷却至室温后,加入20 mL3%磺基水杨酸溶液并用水定容至100 mL,充分混匀后,静置15 min。溶液经滤纸过滤,滤液依据标准曲线的线性范围确定稀释倍数,备用。
1.2.2.3 净化 净化柱ON GUARD Ⅱ RP柱的活化按照以下步骤操作:将ON GUARD Ⅱ RP柱使用前依次用10 mL甲醇、15 mL超纯水进行活化,活化液体流速控制在3 mL/min,放平,静置20 min后使用。
取上清液依次通过0.45 μm水相滤膜和净化柱,弃去前面3 倍柱体积洗脱液,收集后面的洗脱液待测。
1.2.3 离子色谱条件 赛默飞CarboPacTM PA20色谱柱(4.0 mm×150 mm,10 μm),CarboPacTM PA20 保护柱(4.0 mm×50 mm,10 μm),流动相:A为去离子水、B为200 mmol/L氢氧化钠溶液;流速:0.4 mL/min,进样体积:10 μL,柱温:30 ℃(表1)。
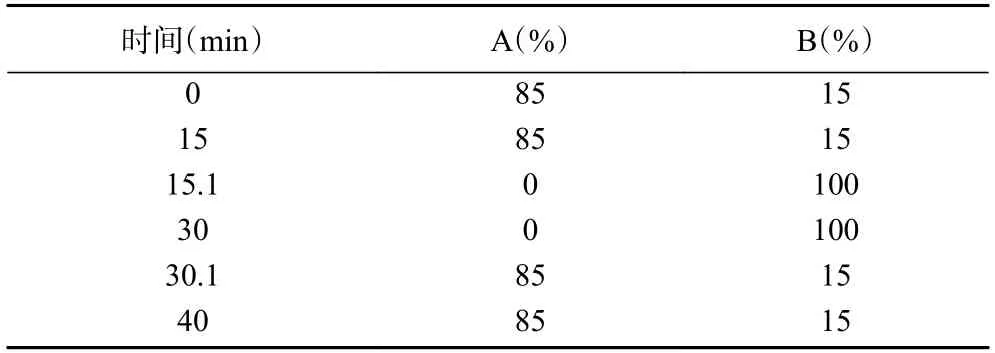
表1 梯度洗脱程序Table 1 The program of gradient elution
1.3 数据处理
色谱结果积分处理采用Thermo Chromenleon 6.8 SR14 色谱工作站,并利用Excel软件进行数据统计及数据分析。
2 结果与讨论
2.1 样品前处理的优化
食品中基质复杂,不仅会对目标色谱峰产生干扰,而且容易对色谱柱及检测器造成损坏。因此,样品前处理过程中使用了净化柱,目的是去除试样溶液中的色素、少量的大分子物质、脂类等,以保护分析柱,常用的净化柱有ON GUARD A柱,ON GUARD Na柱,ON GUARD Ⅱ RP柱等。A柱主要用于去除样品溶液中的阴离子污染物并中和强酸;Na柱主要用于去除样品溶液中的金属离子;RP柱主要用于去除样品溶液中的疏水性物质及表面活性剂[33]。考虑到样品中可能存在部分脂溶性物质,本实验选用了ON GUARD Ⅱ RP柱,选取果粒饮料、西梅饮料、酵素、谷物棒、配方奶粉作为实验样品,对ON GUARD ⅡRP柱对目标物的回收率做了考察,异麦芽糖、异麦芽三糖、潘糖的平均回收率分别为99.31%、99.85%、99.54%,回收率均达99%以上,因此实验确定选用该净化柱。
此外,在样品前处理过程中增加除蛋白的步骤以减少对结果的干扰[33]。常见的除蛋白的方法有盐析法[28]、有机试剂沉淀法[28,34]、沉淀剂法[35]、酸沉淀法[36−37]等。本实验从样品类型、色谱柱、淋洗液等综合因素考虑沉淀剂的选择及适用性。盐析法需加入大量盐类物质,上机前需对样品溶液进行脱盐处理,否则高浓度的盐易对色谱峰的峰形及保留时间造成影响。有机试剂沉淀法操作简单,沉淀蛋白的分辨能力高于盐析法,但考虑到乙腈、乙醇等有机试剂挥发性较强,易对环境造成污染,故排除了有机试剂沉淀法。常用的蛋白沉淀剂有亚铁氰化钾-乙酸锌,沉淀效率高,但考虑到金属离子对离子色谱的不良影响,故不考虑沉淀剂法。综上,本实验采用冰醋酸沉淀法,取6 份样品,分别加入0.05 mol/L的冰醋酸水溶液1、2、3、4、5、6 mL,考察蛋白质的沉淀情况,结果表明,加入5 mL0.05 mol/L的冰醋酸水溶液时,样品中的蛋白质完全沉淀,因此,确定加入5 mL 0.05 mol/L的冰醋酸水溶液作为蛋白沉淀剂,沉淀效果较好且对离子色谱分析无影响。
2.2 色谱柱的选择
实验采用阴离子交换柱CarboPacTM PA1、PA20,比较两种色谱柱对样品中可能存在的半乳糖、葡萄糖、果糖、乳糖、蔗糖、麦芽糖、异麦芽糖、异麦芽三糖、潘糖的分离效果。实验发现,色谱柱CarboPacTM PA20 树脂基核及覆盖在树脂表面的键合季铵基团的乳胶比比色谱柱CarboPacTM PA1 小、分离速度适中,在170 mmol/L氢氧化钠溶液浓度下,半乳糖、葡萄糖、果糖在色谱柱CarboPacTM PA20 能够得到较好分离。色谱柱CarboPacTM PA1 也可以将9 种糖进行分离,但分离效果较色谱柱CarboPacTM PA20差且分析时间长,当样品中乳糖含量高时分离效果不理想,综上,色谱柱CarboPacTM PA20 相较而言具有更大优势,因此本实验采用色谱柱CarboPacTM PA20 进行相关测定。其中,色谱柱CarboPacTM PA20 色谱图和色谱柱CarboPacTM PA1 色谱图见图1 和图2。

图1 CarboPacTM PA20 色谱柱色谱图Fig.1 The chromatogramof CarboPacTM PA20 column

图2 CarboPacTM PA1 色谱柱色谱图Fig.2 The chromatogramof CarboPacTM PA1 column
2.3 流动相的选择
考察了不同浓度的氢氧化钠溶液作为流动相的色谱分离条件,结果表明,流动相中氢氧化钠溶液浓度的高低直接影响分离效果和分离时间,提高氢氧化钠溶液的浓度能够缩短出峰时间,但是可能造成分离度下降,降低氢氧化钠溶液浓度可以增加分离度,同时使分离时间延长,难以在较短的时间内使目标物得到较好的分离度,且半乳糖、葡萄糖、果糖、蔗糖、乳糖等洗脱需要的氢氧化钠溶液较异麦芽三糖、潘糖洗脱需要的氢氧化钠溶液低,综合以上因素考虑,采用梯度洗脱的方式,分离度和分离时间结果满意,样品测定效果较好。当样品中乳糖含量较高时,可以通过酶解法酶解乳糖或者调整梯度洗脱程序使异麦芽糖和乳糖实现较好的分离,样品色谱图如图3 所示,酶解乳糖后产生半乳糖和葡萄糖,样品色谱图如图4所示。

图3 样品色谱图Fig.3 The chromatogram of sample

图4 样品色谱图Fig.4 The chromatogram of sample
2.4 方法线性回归方程
以目标物质量浓度(mg/L)为横坐标,峰面积为纵坐标,绘制标准曲线,得到回归方程和相关系数。结果表明,异麦芽糖、异麦芽三糖、潘糖在0.8~16.0 mg/L范围内线性关系良好,各化合物线性回归方程、相关系数见表2。

表2 3 个化合物的回归方程、相关系数Table 2 Regression equations and correlation coefficients of the three compounds
2.5 方法的定量限
当进样量为10 μL时,将异麦芽糖、异麦芽三糖、潘糖的色谱信号峰与基线信号进行比较,以信噪比约为10 的浓度进行定量限计算,得异麦芽糖、异麦芽三糖、潘糖的定量限均为0.01 g/100 g。
2.6 精密度的测定
平行称取6 份果味饮料,按照前处理方法进行提取并上机测定。结果如表3 所示,异麦芽糖、异麦芽三糖、潘糖的相对标准偏差(RSD)分别为3.42%、2.34%、1.11%,均小于5%,精密度良好。

表3 精密度实验结果(g/100 g)Table 3 The results of precision (g/100 g)
2.7 回收率的测定
平行准确称取6 份果味饮料,按照样品前处理方法进行提取并上机测定,取六次测定结果的平均值为目标物的本底值;准确称取9 份1 g(精确至0.0001 g)果味饮料分别加入不同量的低聚异麦芽糖,按照样品前处理方法进行提取并上机测定,结果见表4。结果显示,三种低聚异麦芽糖的回收率在85.63%~105.22%之间,相对标准偏差在2.48%~4.49%之间,方法回收率良好,准确度较高。

表4 回收率实验结果Table 4 The results of recovery
回收率公式如下:P=(X1−X0)/ m×100
式中,P:加入的标准物质的回收率;m:加入的标准物质的量;X1:加标试样的测定值;X0:本底的测定值。
2.8 样品测定结果
本次实验共收集了果味固体饮料(饮料1)、西梅饮料(饮料2)、果粒饮料(饮料3)、乳酸菌饮料(饮料4)、保健果茶、蜂蜜、酵素、果冻、营养奶昔、功能食盐、苏打饼干、谷物棒、配方奶粉等13 种不同类型的产品,按照已建立的方法测定样品中6 种单双糖和三种低聚异麦芽糖的含量,结果见表5。该方法操作简单,可行性好,专一性强,适合不同类型食品中低聚异麦芽糖的测定。

表5 不同食品中糖的含量(g/100 g)Table 5 Sugars concentration detected in different of foods(g/100 g)
3 结论
本文用离子色谱-脉冲安培检测方法同时测定食品中半乳糖、葡萄糖、果糖、乳糖、蔗糖、麦芽糖、异麦芽糖、异麦芽三糖、潘糖,并对样品的前处理及色谱柱和流动相的选择进行了考察和优化、确定了梯度洗脱程序,使其可用于多种类型的食品中的九种糖的检测。结果表明,该方法在0.8~16.0 mg/L范围内线性关系良好,相关系数r均大于0.999,异麦芽糖、异麦芽三糖、潘糖精密度RSD分别为3.42%、2.34%、1.11%,回收率在85.63%~105.22%之间,相对标准偏差在2.48%~4.49%之间。方法前处理简单、分离效果好、专一性强、结果准确度高、精密度好。该方法可以为测定多种类型食品中的低聚异麦芽糖的检测提供较为准确、可靠的分析方法,为监管政策的落实和实施提供必要的技术支持和有力保障,具有较高的实际应用价值。
