‘六月早’蜜柚叶绿体基因组及其特征分析
2021-07-20徐世荣陈燕琼潘东明潘鹤立
徐世荣 陈燕琼 潘东明 潘鹤立
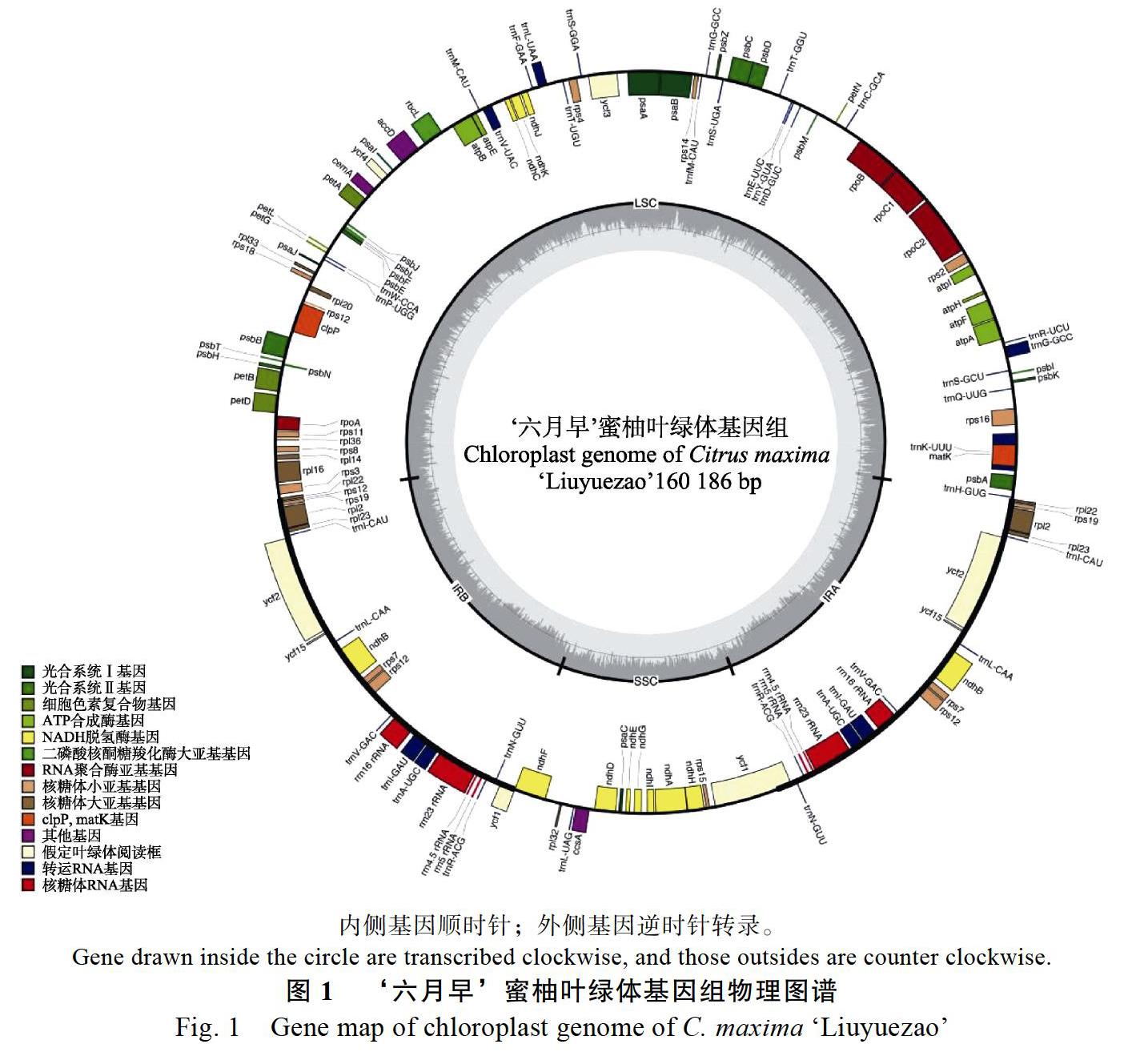
摘 要:‘六月早蜜柚(Citrus maxima ‘Liuyuezao)为国内重要的柚品种琯溪蜜柚早熟芽变品种。以‘六月早蜜柚为材料,利用二代测序进行全基因组重测序,从中组装获取叶绿体基因组并对其进行注释。结果表明:组装获得的‘六月早蜜柚的叶绿体基因组全长160 186 bp,四分体结构由大单拷贝区(large single copy, LSC)、小单拷贝区(small single copy, SSC)和反向重复区(inverted repeat, IR)组成,3个分区的长度分别为87 939、18 395、26 926 bp。注释得到133个基因,其中包含89个编码基因,37个tRNA和8个rRNA。共识别到31个短串联重复序列,101个SSR位点。将已公开发表的29个芸香科叶绿体基因组使用最大似然法进行系统发育关系的构建,结果表明,‘六月早蜜柚与甜橙(C. sinensis)、柠檬(C. limon)和C. platymamma的亲缘关系较近。
关键词:琯溪蜜柚;叶绿体基因组;SSR;系统发育
中图分类号:S961.6 文献标识码:A
Abstract: Citrus maxima ‘Liuyuezao is a precocious bud mutation of Guanximiyou, an important pomelo variety of China. In this study, the whole genome resequencing of C. maxima ‘Liuyuezao was conducted using next-generation sequencing. The chloroplast (cp) genome was assembled and annotated. The cp genome had a total length of 160 186 bp, the tetrad structed was consisted with large single copy (LSC), small single copy (SSC) and inverted repeat (IR), with the length in 87 939 bp, 18 395 bp and 26 926 bp respectively. The cp genome contained a total of 133 genes, including 89 protein-coding genes, 37 tRNA genes and 8 tRNA genes. We identified 31 tandem repeat and 101 SSRs. Combined with 29 published cp genome sequences of Rutaceae, we used the maximum likelihood method to reconstruct the phylogenetic relationship of Rutaceae. The result showed that C. maxima ‘Liuyuezao had a close relationship with C. sinensis, C. limon and C. platymamma.
Keywords: Guanximiyou; chloroplast genome; SSR; phylogeny
DOI: 10.3969/j.issn.1000-2561.2021.05.004
叶绿体在维持地球生命中起着至关重要的作用,通过光合作用和氧气再释放将太阳能转化为碳水化合物。叶绿体基因组编码许多参与光合作用和其他代谢过程的关键蛋白质[1]。在被子植物中,叶绿体为母系遗传,且具有结构稳定、序列高度保守和遗传重组率低等特点,被广泛应用于重建系统发育关系,揭示物种内部和物种之间的巨大差异[2–4]。对叶绿体基因组数据的了解可提高我们对植物生物学和多样性的认识。
柚[Citrus maxima (Burm.) Merr.]是我国特产果树,为芸香科(Rutaceae)柑橘属(Citrus)常绿乔木果树,而柑橘属水果是世界上种植最广泛的水果之一,与苹果、葡萄、香蕉并称为世界四大水果。柚作为一个古老的品种,在历史的繁衍进化过程中通过自身芽变或者与其它柑橘杂交产生了许多果实大小和酸度不同的新品种,如葡萄柚、酸橙等,极大的丰富了柑橘家族[5]。国内的柚类品种主要为琯溪蜜柚、沙田柚、四季柚、度尾文旦、强德勒柚、坪山柚和暹罗柚等。其中栽培面积最大的品种为福建平和的琯溪蜜柚,仅福建地区产量就约占全国柚类总产量的2/3、世界的1/3。
琯溪蜜柚因其果大皮薄、肉嫩汁多无核、营养物质丰富及具有独特的香气而广受消费者的喜爱,目前产品不仅占据了国内的大小商场,还远销至欧盟、美国、加拿大等国家。近10多年已逐渐发展成为柑橘类果树中最具市场竞争力和发展潜力的水果之一。但其成熟期在每年的10—12月,上市的时间过于集中,导致货架长时间空缺,因此培育不同成熟的琯溪蜜柚是市场的需求。福建平和果农于2007年发现了1株琯溪蜜柚发生了早熟芽变,经过对母树及突变枝嫁接子代的连续多年观察,发现其性状稳定,成熟期比琯溪蜜柚早约45 d,由于在農历6月就能成熟,故命名为‘六月早。
本研究利用高通量测序技术对‘六月早蜜柚进行测序,以已发表的柚的叶绿体基因组为参考,获取‘六月早蜜柚的叶绿体基因组序列,进行注释和序列特征分析,并与其他的芸香科的物种进行比较分析和系统发育关系推断,以期为芸香科的分子标记开发和系统发育研究提供参考。
1 材料与方法
1.1 材料
以‘六月早琯溪蜜柚为材料,其幼嫩的叶片组织采于福建平和县小溪镇果园。
1.2 方法
1.2.1 总DNA的提取和测序 利用TIANGEN试剂盒提取‘六月早琯溪蜜柚的总DNA,用琼脂糖凝胶电泳确定总DNA具有较好的完整性,并用分光光度计检测DNA的质量。检测的DNA浓度>50 ng/μL,无色素污染、无明显RNA、无蛋白质等杂质污染的样品视为合格样品。将检测合格的DNA样品送至深圳华大基因科技服务有限公司进行文库的构建,最后在Illumina HiSeq平台上机测序,获得质控过滤后的Clean data 12 Gb的数据量,覆盖该物种测序深度37×。
1.2.2 叶绿体基因组的组装与注释 对下机的数据进行过滤,包括去除低质量的序列和接头。从GenBank中下载已发表的柚叶绿体基因组序列(GenBank登录号:KY055833.1)作为参考序列,使用GetOrganelle toolkit[6]初步组装出叶绿体基因组的序列,再使用Bandage[7]根据叶绿体基因组的机构特征手动拼接出完整的质体基因组。
使用Geneirous primer v2019.03[8]对组装好的序列进行注释,注释使用已发表的柚作为注释参考,再人工校正基因的起始和终止密码的位置。序列的注释可视化在OrganellarGenomeDRAW(OGDRAW)v1.3.1(https://chlorobox.mpimp-golm. mpg.de/OGDraw.html)进行[9]。将组装注释好的‘六月早蜜柚的叶绿体基因组上传至GenBank数据库,获得登录号:MT527726。
1.2.3 叶绿体基因组的特征分析 串联重复序列的识别用Tandem repeat finder软件[10],使用默认参数。简单重复序列(simple sequence repeat,SSR)的识别使用Perl脚本MISA(microSAtellite identification tool),参数设置为单核苷酸(mononucleotide)、二核苷酸(dinucleotide)、三核苷酸(trinucleotide)、四核苷酸(tetranucleotide)、五核苷酸(pentanucleotide)和六核苷酸(hexanucleotide)的重复数阈值分别为10、5、4、3、3和3。使用在线分析软件CodonW 1.4.4(https:// galaxy.pasteur.fr/?form=codonw)对叶绿体基因组内的编码基因进行密码子的使用偏好性分析。
1.2.4 芸香科物种叶绿体基因组比较分析 从GenBank数据库中下载柠檬(Citrus limon)、山小橘(Glycosmis pentaphylla)、假黄皮(Clausena excavata)、芸香(Ruta graveolens)、花椒(Zanthoxylum bungeanum)、黄檗(Phellodendron amurense)、香肉果(Casimiroa edulis)等29个芸香科隶属于11个属的其他物种的叶绿体基因组。每个属选1个代表种共11个种进行边界的扩张与收缩分析,使用在线程序Irscope(https:// irscope.shinyapps.io/irapp/)对IR、SSC和LSC区域进行边界变化分析[11]。
以无患子科(Sapindaceae)栾树(Koelreuteria paniculata)为外类群,与其他29个芸香科物种进行系统发育树的构建。将30个叶绿体基因组序列使用Homblock软件进行同源共线性的比对[12],识别出的共线性区域用于系统发育树的构建。使用IQ-tree对数据集进行系统发育树的构建[13]。
2 结果与分析
2.1 ‘六月早蜜柚叶绿体基因组的基本特征
‘六月早蜜柚的叶绿体基因组结构为经典四段式结构,包含大单拷贝区(large single copy,LSC),2个反向重复区(inverted repeat,IR)和小单拷贝区(small single copy,SSC)(圖1)。叶绿体基因组的总长度为160 186 bp,GC含量为38.5%,其中LSC区长87 939 bp,IR区长26 926 bp,SSC区长18 395 bp。共有基因133个,包括编码基因89个,tRNA 37个,rRNA8个。其中20个基因含有2个拷贝,包括8个蛋白编码基因(rpl2、rpl23、rps7、rps19、ndhB、ycf1、ycf2和ycf15),8个tRNA(trnA-UGC、trnG-GCC、trnI-CAU、trnI-GAU、trnL-CAA、trnN-GUU、trnR-ACG和trnV-GAC)和4个rRNA(rrn4.5、rrn5、rrn16和rrn23)。
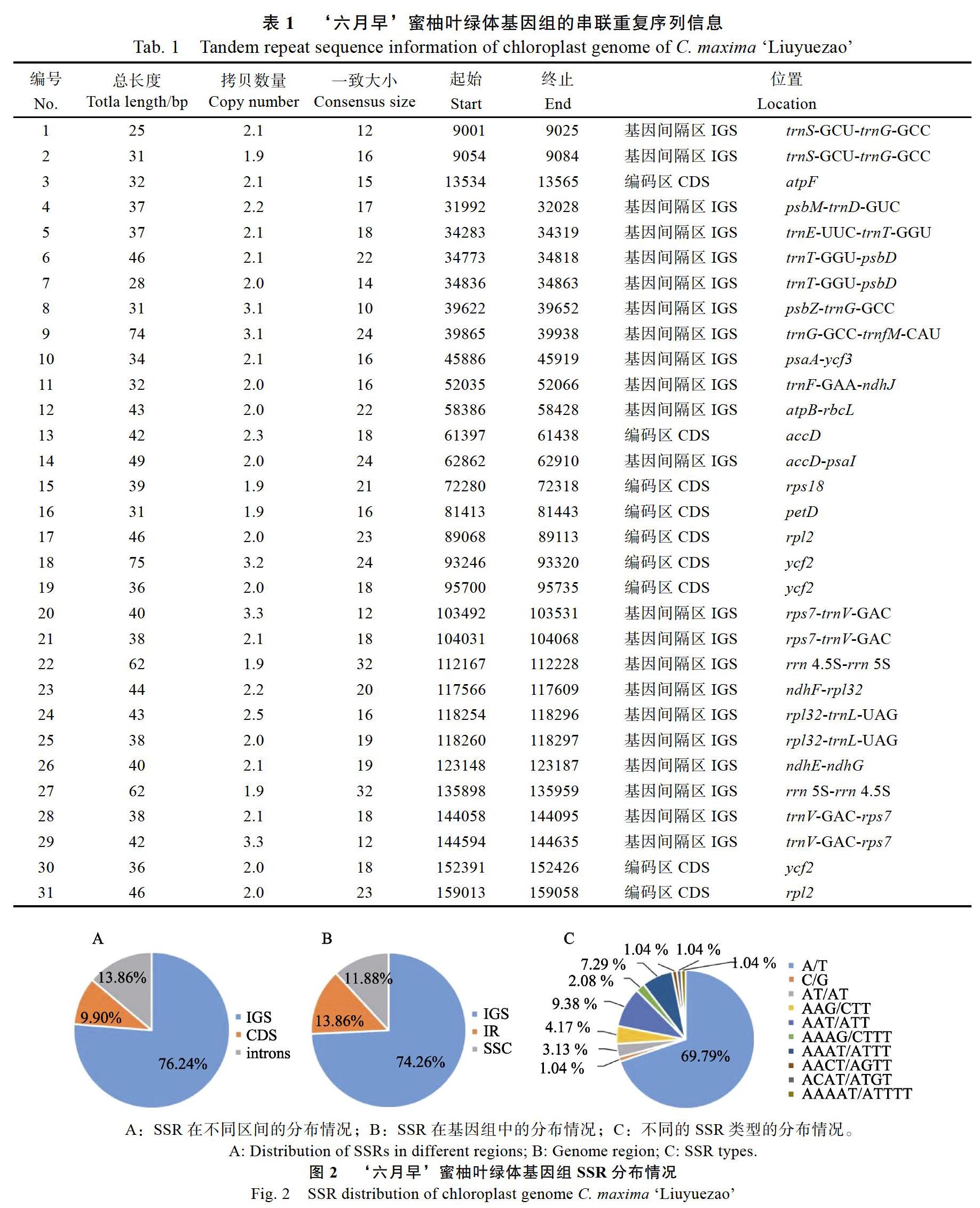
2.2 ‘六月早蜜柚叶绿体基因组的重复序列
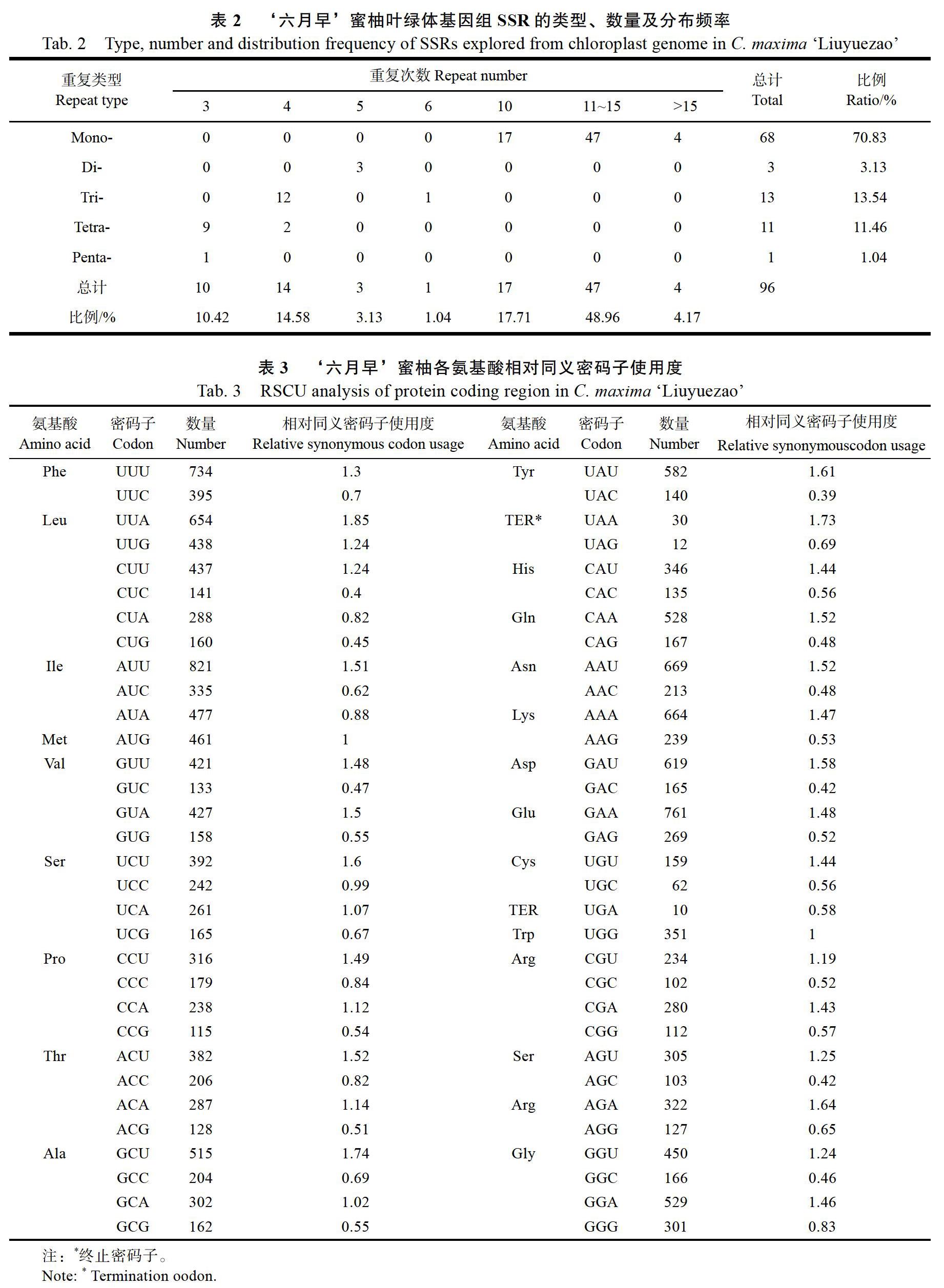
识别叶绿体基因组的重复序列,特别是SSR对于分子标记的开发,有助于为物种的遗传发育及分子育种研究提供参考。对‘六月早的重复序列进行检测,共识别出31个短串联重复序列的区域,绝大部分分布在基因间隔区(IGS),长度范围在25~75 bp之间,占基因组序列的0.81%(表1)。共检测到101个SSR,其中包括5个复合型SSR、68个单碱基重复、3个二碱基重复、13个三碱基重复、11个四碱基重复和1个五碱基重复(表2,图2)。SSR序列长度为10~35 bp,大部分SSR均分布在基因组的大单拷贝区(占74.26%)和基因间隔区(占76.34%,图2)。
2.3 ‘六月早蜜柚叶绿体基因组的密码子偏好性
密码子是基因编码区蛋白翻译的核心元件,分析编码区密码子的使用特征,对基因功能和系统进化研究具有重要意义,为后续研究提供基础资料。将‘六月早蜜柚叶绿体基因组中得到的88个编码基因(coding DNA sequence,CDS)用于分析,为了保证结果的准确性,去除了重复的基因序列和长度小于300 bp序列,将剩下的52条CDS用于密码子的偏好性分析。RSCU值大于1.00的密码子为30个,其中29个以A或U结尾,1个以G或C结尾(表3),表明这些密码子偏向A/U结尾,‘六月早蜜柚与大多数被子植物的密码子偏好性类似。
2.4 IR边界的收缩与扩张
选择了11个物种代表芸香科的11个属进行IR边界的比较分析。从图3可见,芸香科植物具有相似的基因结构和基因顺序,差异主要发生在JLB(LSC与IRb边界)和JSB(IRb与SSC边界)边界。在JLB边界表现出2种类型,第1种类型为无基因横跨边界,仅见黄皮属的假黄皮Clausena excavata和小芸木属的Micromelum minutum,位于LSC的rps3为离边界最近的基因分别距JLB 38 bp和115 bp;第2种类型是rpl22基因横跨了JLB边界,其余9个种均为这种类型,其中跨到LSC边界的长度为67~295 bp。JSB边界则表现出了3种类型,第1种类型为无基因处于边界上,仅表现在酒饼簕属广东酒饼簕(Atalantia kwangtungensis)中;第2种类型为ycf1基因横跨JSB边界,在黄檗属黄檗(Phellodendron amurense)、花椒属(Zanthoxylum tragodes)、芸香属芸香(Ruta graveolens)和‘六月早蜜柚4个种中表现有1~35 bp跨到SSC区;第3种类型为ndhF基因横跨JSB边界,其余的6个种、芸香和‘六月早蜜柚有1~31 bp跨越IRa区域。JLA边界也表现出3种类型,表现为不同的种在边界处trnH和rpl22基因的位置不同,第1种为边界无基因;第2种为trnH跨越边界;第3种为rpl22跨越边界。JSA则非常保守,均为ycf1基因跨越了边界,有1080~1130 bp位于IRa。
2.5 系统发育分析
为了确定‘六月早蜜柚在芸香科中的进化地位,基于叶绿体基因组序列构建29个的芸香科物种系统发育关系,其中柑橘属10种,酒饼簕属1种,Merrillia属1种,山小橘属2种,小芸木属1种,咖喱树属1种,黄皮属1种,芸香属1种,花椒属9种,黄檗属1种和香肉果属1种,以无患子科栾树为外类群。由图4可知,芸香科由两大进化支组成,各分支均获得很高的自展值(BSML均为100);花椒属、黄檗属和香肉果属聚为一支,为最先分化的分支,且均属于Amyridoideae亚科;柑橘属、酒饼簕属、Merrillia属、山小橘属、小芸木属、咖喱树属、黄皮属和芸香属聚为一支,其中芸香属为最先分化的属,其他的7个属聚为一支;‘六月早蜜柚与其他柑橘属聚为一支,与Citrus platymamma、柠檬和甜橙分支成姐妹关系。
3 讨论
‘六月早蜜柚同其他被子植物一样,具有四分结构环状的叶绿体基因组,其基因顺序、GC含量也同其他柑橘属的植物基本一样[14–16],这也说明了在柑橘属内叶绿体基因组非常保守。植物的叶绿体基因组长度在100~220 kb之间,芸香科内叶绿体基因组长度在157 434 bp(芸香)~161 172 bp(假黄皮)之间,而差异主要表现在反向重复(IR)区的收缩、扩张或缺失,因此,IR区对于叶绿体基因组的结构稳定性和决定叶绿体基因组大小方面的作用不可忽视[17-18]。重复序列的存在从原核生物到真核生物的进化过程中呈递增趋势,具有保护编码基因和作为进化的动力促进形成新基因的作用,是进化的必然结果[19]。‘六月早蜜柚的重复序列大部分分布于IGS区,且超过一半(16/31)分布于LSC区,这表明重复序列对该物种不具有特定功能。SSR广泛存在于基因组中,长度在1~6 bp短的重复基序(motif),具有高多态性、多等位性、共显性等特点,同时因其具有良好的种间甚至种内的遗传变异区分精度,一直被广泛用于植物资源研究[20–23],本研究从‘六月早蜜柚叶绿体基因组中识别到的SSR可提供该品种的种间多态性,为柑橘属品种间的亲缘关系和分子鉴定提供可靠的遗传标记。
本研究将已公开发表的所有芸香科植物的叶绿体基因组从GenBank中获取,包含29个种,隶属于11個属,3个亚科,对其进行芸香科的系统发育关系的重建。在前人的研究中,仅使用叶绿体基因片段进行芸香科的构建,如使用rps16内含子和trnL-trnF基因间隔区对65个芸香科物种进行分析,发现亚科与族的关系都需要重新评估[24]。本研究结果获得了稳固的系统发育关系,各亚科和各属的单系性均得到了很好的支持,其中柑橘亚科与芸香亚科成姐妹关系,构成的分支与最早分化的脂檀亚科成姐妹关系。但由于本研究使用的物种序列有限,构建的系统发育关系仍不够全面,有待进一步完善。
参考文献
[1] Daniell H, Lin C S, Yu M, et al. Chloroplast genomes: diversity, evolution, and applications in genetic engineering[J]. Genome Biology, 2016, 17(1): 134.
[2] Jansen R K, Cai Z, Raubeson L A, et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns[J]. Proceedings of the National Academy of Sciences, 2007, 104(49): 19 369-19 374.
[3] Song Y, Yu W B, Tan Y, et al. Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the Magnoliids[J]. Genome Biology and Evo-lution, 2017, 9(9): 2354-2364.
[4] Gao C, Deng Y, Wang J. The complete chloroplast genomes of Echinacanthus species (Acanthaceae): phylogenetic relationships, adaptive evolution, and screening of mo-lecular markers[J]. Frontiers in Plant Science, 2018, 9: 1989.
[5] Wu G A, Terol J, Ibanez V, et al. Genomics of the origin and evolution of Citrus[J]. Nature, 2018, 554(7692): 311-316.
[6] Jin J J, Yu W B, Yang J B, et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes[J]. Genome Biology, 2020, 21(1): 1-31.
[7] Wick R R, Schultz M B, Zobel J, et al. Bandage: interactive visualization of de novo genome assemblies[J]. Bioinformatics, 2015, 31(20): 3350-3352.
[8] Kearse M, Moir R, Wilson A, et al. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data[J]. Bioinformatics, 2012, 28(12): 1647-1649.
[9] Greiner S, Lehwark P, Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes[J]. Nucleic Acids Research, 2019, 47(W1): 59-64.
[10] Benson G. Tandem repeats finder: a program to analyze DNA sequences[J]. Nucleic Acids Research, 1999, 27(2): 573-580.
[11] Amiryousefi A, Hyv?nen J, Poczai P. Irscope: an online program to visualize the junction sites of chloroplast genomes[J]. Bioinformatics, 2018, 34(17): 3030-3031.
[12] Bi G, Mao Y, Xing Q, et al. Homblocks: a multiple-alignment construction pipeline for organelle phylogenomics based on locally collinear block searching[J]. Genomics, 2018, 110(1): 18-22.
[13] Nguyen L T, Schmidt H A, Von Haeseler A, et al. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies[J]. Molecular Biology and Evolution, 2015, 32(1): 268-274.
[14] Bausher M G, Singh N D, Lee S B, et al. The complete chloroplast genome sequence of Citrus sinensis (L.) Osbeck var ‘Ridge Pineapple: organization and phylogenetic relationships to other angiosperms[J]. BMC Plant Biology, 2006, 6(1): 725-730.
[15] Su H J, Hogenhout S A, Al-Sadi A M, et al. Complete chloroplast genome sequence of Omani lime (Citrus aurantiifolia) and comparative analysis within the rosids[J]. PLoS One, 2014, 9(11): 113049.
[16] Xu S R, Huang C Y, Deng Y T, et al. The complete chloroplast genome sequence of Citrus maxima (Burm.) Merr. ‘Guanximiyou[J]. Mitochondrial DNA Part B, 2020, 5(1): 482-483.
[17] Palmer J D, Thompson W F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost[J]. Cell, 1982, 29(2): 537-550.
[18] Wang R J, Cheng C L, Chang C C, et al. Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots[J]. BMC Evolutionary Biology, 2008, 8(1): 1-14.
[19] 艾對元. 基因组中重复序列的意义[J]. 生命的化学, 2008(3): 343-345.
[20] Provan J, Powell W, Hollingsworth P M. Chloroplast microsatellites: new tools for studies in plant ecology and evolution[J]. Trends in Ecology & Evolution, 2001, 16(3): 142-147.
[21] Kantety R V, La Rota M, Matthews D E, et al. Data mining for simple sequence repeats in expressed sequence tags from barley, maize, rice, sorghum and wheat[J]. Plant Molecular Biology, 2002, 48(5-6): 501-510.
[22] Lassois L, Denancé C, Ravon E, et al. Genetic diversity, population structure, parentage analysis, and construction of core collections in the French apple germplasm based on SSR markers[J]. Plant Molecular Biology Reporter, 2016, 34(4): 827-844.
[23] Singh G, Aulakh P S, Sarao N K, et al. Molecular verification of putative zygotic seedlings in different intra-specific crosses in Mandarins (Citrus reticulata) by SSR markers[J]. Agricultural Eesearch, 2019, 8(1): 21-26.
[24] Groppo M, Pirani J R, Salatino M L F, et al. Phylogeny of Rutaceae based on twononcoding regions from cpDNA[J]. American Journal of Botany, 2008, 95(8): 985-1005.
责任编辑:黄东杰
