地下水氯代烃污染修复技术研究进展
2021-07-17任加国郜普闯徐祥健尚长健姜永海
任加国, 郜普闯,, 徐祥健, 夏 甫, 韩 旭, 尚长健, 生 贺, 杨 昱*, 姜永海
1.山东科技大学地球科学与工程学院, 山东 青岛 266590
2.中国环境科学研究院, 环境基准与风险评估国家重点实验室, 北京 100012
3.中国环境科学研究院, 国家环境保护地下水污染模拟与控制重点实验室, 北京 100012
氯代烃作为一种常用的化工原料,在工业清洗领域以及衣服脱脂领域发挥着重大作用[1]. 它常常通过工业废水和生活污水的不合理排放、废弃物堆积场地渗滤液的渗漏、有毒有害化学废物的不正当泄漏等方式进入到地下水,污染地下水环境[2]. 同时,大部分氯代烃具有致癌、致畸、致突变的潜在毒性,对人类身体健康及生产生活也造成了极大的威胁[3]. 因此,美国、欧盟以及我国都将其作为优先控制的有毒有害有机污染物之一.
近年来,伴随工业化进程的不断推进,地下水中氯代烃污染形势日益严峻. 北京、上海、济南等地的部分化工场地以及鲁北平原、珠三角地区等区域地下水均受到不同程度的污染,且其污染范围不断扩大,并呈现由点向面演变、由局部向区域扩散、由东部向西部扩展、由城市向农村蔓延的趋势[4-8]. 因此,污染场地地下水氯代烃的污染防控刻不容缓. 要想实现对污染场地地下水氯代烃的有效治理就必须掌握场地地下水污染的状况,而场地的污染状况与氯代烃在地下水环境中的赋存状态与行为归趋紧密相关[9]. 基于此,充分了解地下水中氯代烃多相态、多介质赋存状态与迁移转化特征,对其修复治理具有重要意义. 同时,科学高效的污染防治技术的实施是实现地下水中氯代烃绿色高效修复的关键. 目前,国内外专家学者基于对氯代烃不同赋存形态下的迁移转化特征的深入研究,提出了一系列地下水氯代烃污染修复技术,如气相抽提(SVE)技术、热处理强化(TE)技术[10]、多相抽提(MPE)技术、原位化学氧化(ISCO)技术、可渗透反应墙(PRB)技术[11]、原位生物修复(ISB)技术[12]、表面活性剂强化抽出处理(SEAR)技术[13]、监控自然衰减(MNA)技术[14]等.
综上,该文主要从氯代烃在包气带及含水层介质中的赋存状态与迁移转化特征出发,分别阐述了不同赋存状态下最适用的修复技术,并对其技术原理、优缺点及研究进展等进行分析整理,提出地下水氯代烃污染修复研究的展望,为实际污染场地的氯代烃修复工程提供理论参考.
1 氯代烃赋存状态与迁移转化特征
氯代烃属于非水相液体(NAPLs)污染物,难溶于水,相较于水溶性污染物,其在地下水环境中的赋存状态与迁移转化过程更复杂. 充分理解氯代烃在地下水环境中不同赋存状态以及具体迁移转化特征,对于污染场地修复技术的选择具有指导意义.
1.1 地下水环境氯代烃的赋存状态
氯代烃在地下水环境中主要有4种赋存状态:气相、残留相、溶解相和自由相[15],其中地表污染源泄露进入到包气带中的氯代烃为自由相氯代烃,此状态的氯代烃既可以在重力作用及含水层介质的毛细作用下不断向下迁移,也可以在水力梯度下沿水平方向运移,在迁移过程中使污染的范围不断扩大,因此是主要的污染源. 迁移过程中,会有一部分自由态的氯代烃残留在运移路径上,受吸附作用和毛细作用以不连续的薄膜状或液滴状赋存在土壤以及含水层空隙介质或者裂隙介质中,使之无法在重力作用下继续迁移,这部分氯代烃为残留相氯代烃. 当氯代烃继续迁移至地下水面以下,会有一小部分氯代烃作为溶质进入到地下水中并随地下水流不断迁移形成污染羽,这部分氯代烃为溶解相氯代烃;虽然溶解相氯代烃的量比较少,但与人们的联系最紧密,直接影响人们的生产生活与身体健康状况. 最终,自由相氯代烃会在重力作用下滞留在含水层底部并不断累积,形成持久性污染源. 在整个迁移过程中,由于氯代烃为挥发性有机物,因此会有氯代烃不断挥发进入地下水环境,并在浓度梯度下不断迁移,这部分氯代烃称为气相氯代烃. 值得一提的是,包气带滞留一定量残留相与气相氯代烃,可能会在雨水冲刷作用或其他外力条件下继续向下迁移,对地下水造成二次污染,极大地增加了实际场地污染治理难度.
4种赋存状态下的氯代烃并不是独立存在,它们联系紧密,并在一定条件下可以相互转化,具体转化过程见图1.
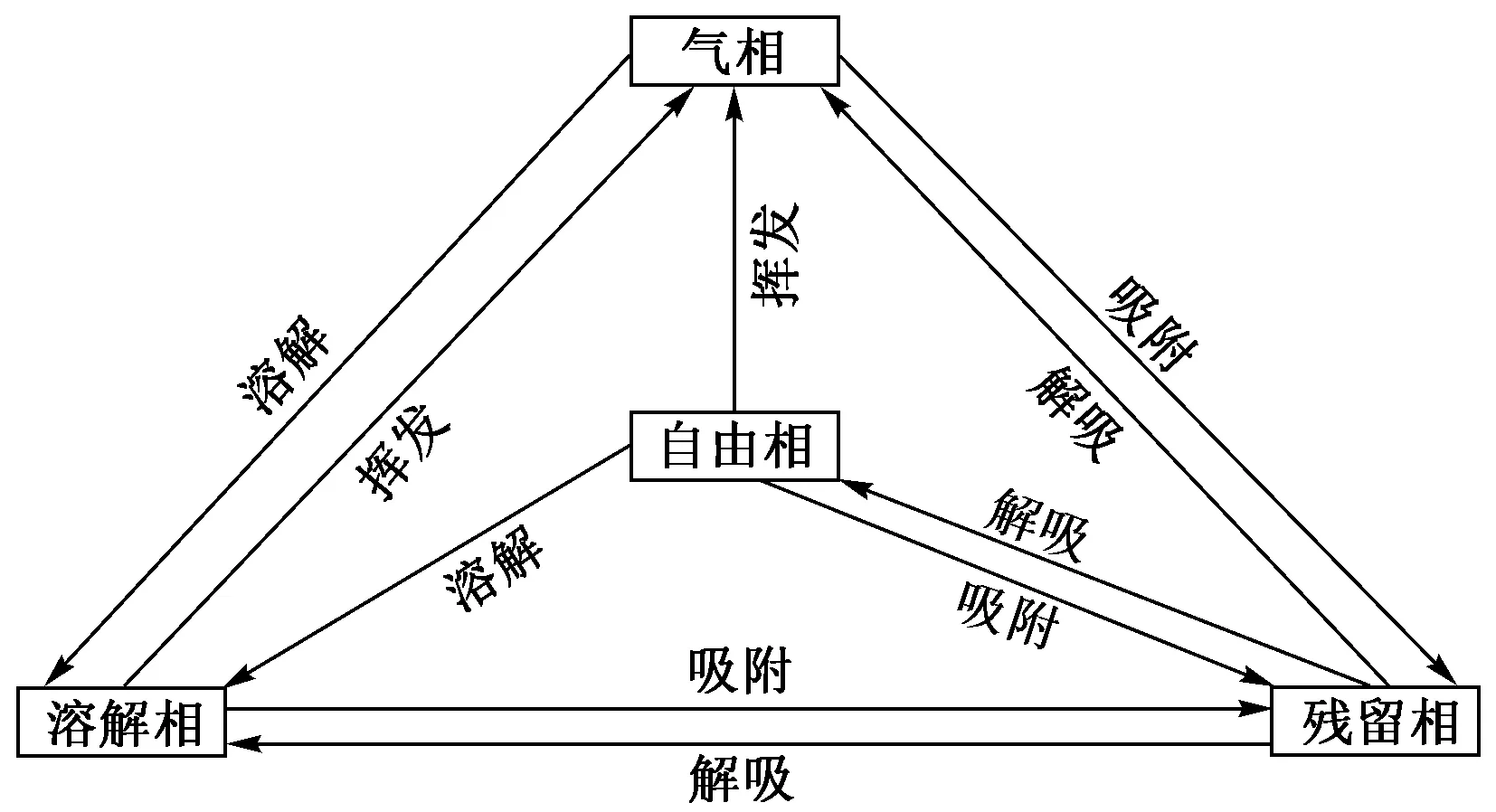
图1 氯代烃4种赋存形态的相互转化
1.2 氯代烃迁移转化影响因素研究
氯代烃的赋存形态多样,其在地下水环境的迁移转化属于多相流问题,不仅受到污染物自身特性的影响,也与所处地下水环境息息相关.
1.2.1氯代烃物理化学性质的影响
不同氯代烃的密度、黏度、溶解度、亨利常数等性质存在一定差异,表现在迁移转化过程中的影响也稍显不同. 现就一些常见氯代烃的性质参数进行汇总(见表1),并对各参数的具体影响进行详细阐述.
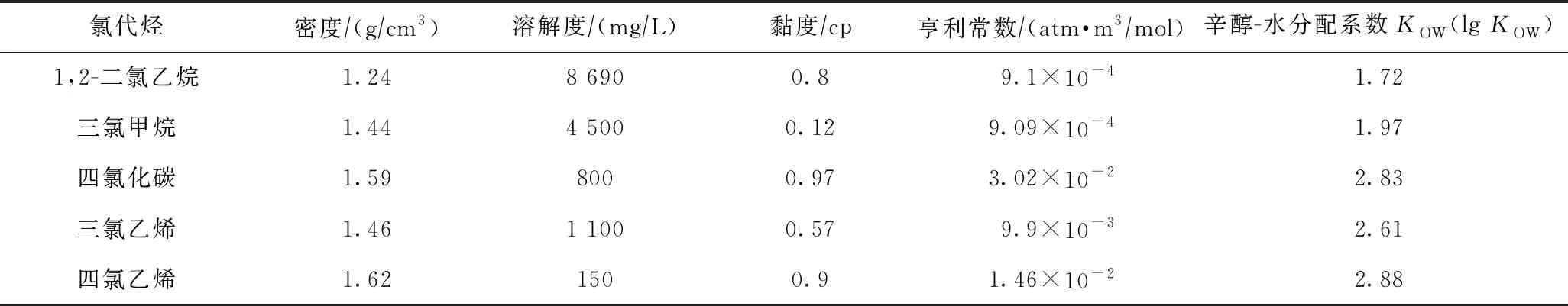
表1 几种常见氯代烃的基本性质参数(20 ℃)
1.2.1.1密度
以水的密度(1 g/cm3)为参考,绝大多数氯代烃密度比水大,为重质非水相液体(DNAPLs),在地下水环境中主要表现为垂向迁移. 同为DNAPL类污染物,高密度氯代烃相较于低密度表现出更深的垂向迁移距离,而低密度氯代烃则表现出更远的水平迁移距离且距离受地下水流速影响较大,这在邓亚平等[16]对饱和均质砂层中3种不同密度DNAPLs运移情况的数值模拟中有所体现.
1.2.1.2溶解度
氯代烃在地下水环境中的溶解是指由其他相向溶解相转化的过程. 1,2-二氯乙烷的溶解度虽远高于三氯甲烷、四氯化碳、三氯乙烯、四氯乙烯等4种氯代烃,但仍属于微溶性物质. 当然,氯代烃的难溶、微溶状态并非不可改变,加入表面活性剂便可以有效增强氯代烃的表观溶解度[17]. 氯代烃增溶的具体机理:①当表面活性剂浓度在超过临界胶束浓度(CMC)后会形成胶束物质,依靠胶束物质的分配作用,氯代烃可以进入胶束物质疏水内核与水的交界面,增强了其在水中的溶解度[18]; ②在一定范围内,随表面活性剂浓度的增大,对氯代烃的增溶效果也逐渐增强. 因而,表面活性剂的使用有利于增强地下水环境中难溶氯代烃抽出处理的效果.
1.2.1.3黏度
黏度是一层流体阻碍另一层流体与其发生相对运动的力,具体到氯代烃主要表现为对氯代烃在地下水环境迁移行为的阻碍作用. 在相同地下水环境条件下,氯代烃黏度越高,其入渗时所需克服的黏滞力就越大,下渗速率就越缓,表现在地下水环境中的下渗距离也就越短,且随氯代烃的不断渗入,会发生水平向扩散,使污染界面变宽. 杨宾等[19]利用二维砂箱模拟多孔介质中柴油、1,2-二氯乙烷和四氯乙烯3种不同黏度NAPLs污染物的迁移特征时也得到了类似结论.
1.2.1.4亨利常数
亨利常数是一种描述氯代烃在气相与液相中分配能力的物理常数. Stupp等[20]研究了甲基叔丁基醚(MTBE)、叔丁醇(TBA)、顺式二氯乙烯(cis-DCE)、氯乙烯(VC)等5种不同亨利常数有机物在水相与气相的分配状态,研究发现,亨利常数较高的cis-DCE和VC容易从水相中脱吸附出来,而MTBE与亨利常数更小的TBA更倾向于在水相中累积,难以从水相中脱吸附出来,因而不能在污水处理厂中将其去除. 因此,不宜用脱吸附方法对低亨利系数氯代烃进行修复.
1.2.1.5辛醇-水分配系数(KOW)
KOW代表了氯代烃在辛醇与水之间的分配方向. 具体来讲,KOW值的大小代表氯代烃受含水层疏水有机物吸附能力的强弱,数值越大,就越容易被疏水性有机物吸附. 大多数氯代烃的lgKOW值较小,因此水溶相氯代烃较不易受吸附作用影响,在含水层中迁移速率较快,从而更容易形成更大范围的污染,这也是地下水中氯代烃类污染物难以彻底清除的原因之一.
1.2.2地下水环境的影响
地下水环境对氯代烃迁移转化的影响主要体现在地下水流速、含水层介质的非均质性以及含水层介质中的生物作用等方面.
地下水流速与含水层介质的非均质性改变了氯代烃在地下水环境的运移特征. 郑菲等[21]在保持其他试验参数相同的条件下,研究了不同流速对四氯乙烯(PCE)运移的影响,结果显示随地下水流速增加,PCE的垂向入渗距离也在不断增加,在流速为1.0 m/d 时,PCE已运移至含水层底部形成了污染池,并且随流速的继续增加,污染池中心也发生相应偏移,可见地下水流速的增加在促进PCE垂向入渗的同时,也促进了PCE的水平运移. 郑菲等[21]同时探究了相同流速下介质非均匀性的影响,发现在均质含水层下规则分布的污染羽形状随含水层介质非均匀性的增加而逐渐变得不规则,其中还可能形成部分的高渗透区与低渗透区,低渗透区的DNAPL运移过程中易发生绕流现象,而高渗透区易发生DNAPL的累积. Page等[22]利用砂箱试验模拟PCE非均相介质下的迁移特征,确定了介质中透镜体对PCE运移与分布的绕流作用.
氯代烃的转化主要由含水层介质中的生物作用完成. Kiecak等[23]利用pH、TOC、氯离子浓度等可用参数构建了污染物迁移转化模型,对波兰东南部地区某水厂受三氯乙烯(TCE)与四氯乙烯(PCE)污染的地下水作了内在生物降解潜力研究. Lukas等[24]对于厌氧地下水环境条件下高氯代烃的降解机制展开了分析,确定了厌氧还原脱氯在其降解过程中的主导作用.
污染物种类与性质的多样性与地下水环境的复杂性,不断改变地下水实际污染状况,加剧了实际污染场地的治理难度.
2 地下水氯代烃修复技术
针对不同赋存状态下氯代烃所造成的污染问题,该文分别综述了几种常用修复技术,并对其技术原理、优缺点以及研究进展作出详细阐述.
2.1 气相氯代烃的修复技术
气相抽提(SVE)技术主要用于修复受挥发性有机污染物污染的土壤以及地下水,因而可以对包气带受气相氯代烃污染的场地实现较好的修复效果,抽提技术示意如图2所示.
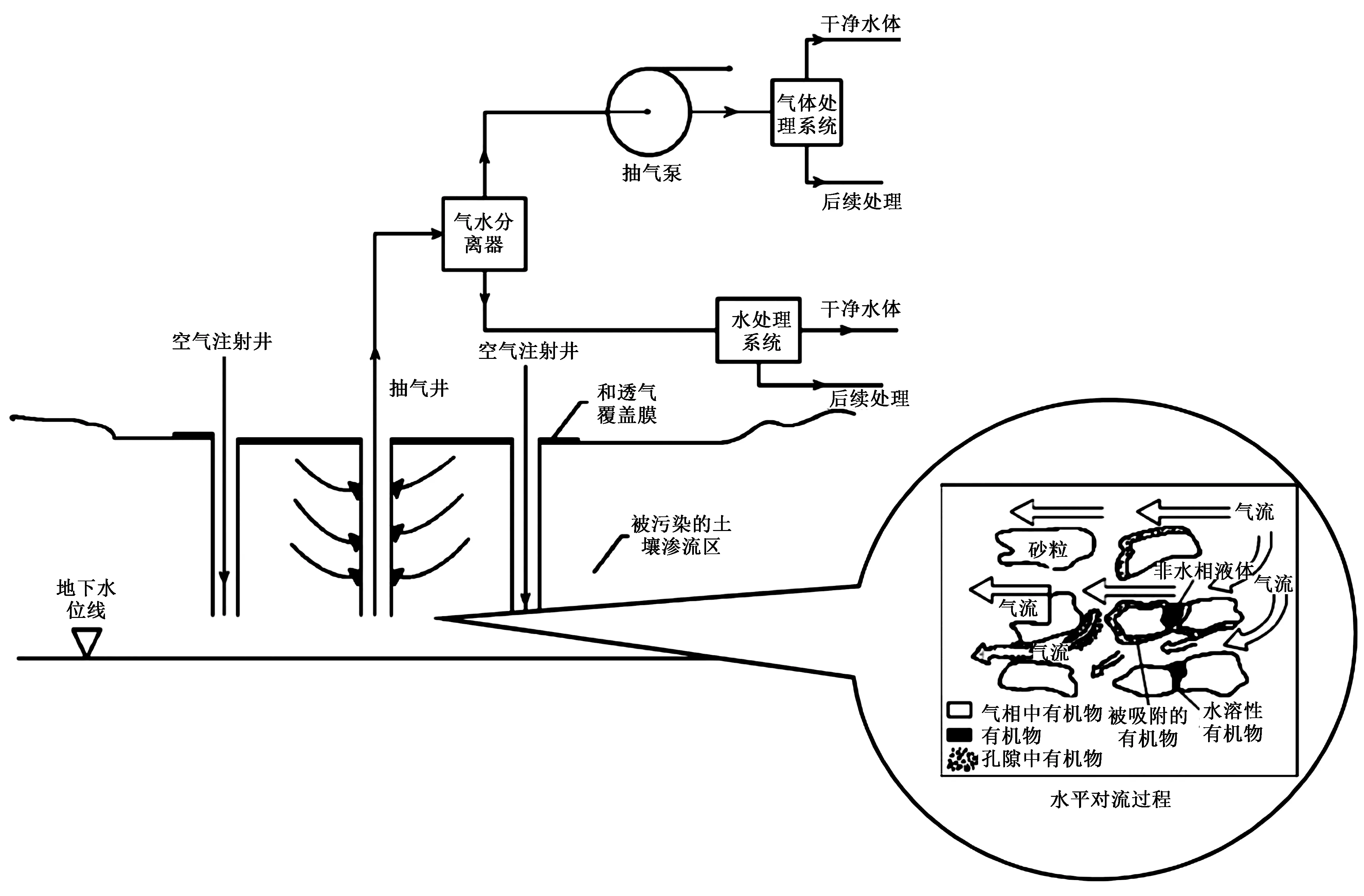
图2 气相抽提技术示意[25]
目前,国内外研究人员对于气相抽提技术的研究已相对成熟. 其中,试验研究主要集中在抽提效率的影响因素分析. 研究[26-28]表明,土壤透气性、含水率、有机质含量及抽气速率均会影响SVE技术的修复效果. 土壤透气性是影响污染物去除效率的关键参数,在渗透率高于10-6cm2的砂土环境中能发挥较优的修复效果[29]. 在试验研究基础上,SVE技术已广泛应用于实际的土壤及地下水修复工程. 在受TCE污染的荷兰某干洗店旧址,通过SVE技术成功将场地土壤污染物的浓度由 4 000 μg/m3降至40 μg/m3[30].
传统SVE技术对场地条件的要求极大地限制了其在复杂场地条件下的应用. 热处理强化(TE)作为一种SVE的强化技术,在场地条件方面表现出了更强的普适性,修复效率也有明显提高,引起了人们的广泛研究[31],逐渐形成电波加热、微波加热、热空气注入、蒸汽注射等热强化方式. 美国孟菲斯一氯代溶剂污染场地,利用原位热强化气相抽提技术对场地8个污染源区进行修复,成功将污染物浓度由 1 000 mg/kg以上降至1 mg/kg以下,且回收了 5 675 kg的氯代溶剂[32]. 但是,热强化方式只适用于污染范围较小的场地,范围过大容易造成修复成本的增加与修复效率的下降;同时,土壤温度的升高,会显著改变土壤的基本性质和生态功能,这也是在TE技术实施过程中值得注意的地方.
2.2 自由相氯代烃的修复技术
自由相氯代烃大多滞留在含水层底部,采用原位修复技术无法有效治理其造成的污染. 而多相抽提(MPE)技术,主要是通过营造真空环境抽取污染场地的部分地下水,使此处的地下水位降低,从而在对流作用下形成地下水降落漏斗,使自由相氯代烃在水力梯度下逐渐向中心汇聚,随后利用泵直接抽取自由相氯代烃至地面并进行相分离与处理,从而实现了对自由相氯代烃的高效修复[33]. 研究发现,MPE技术的修复效果主要受污染场地水文地质条件以及污染物性质的共同影响,表2为MPE技术的适用参数.

表2 MPE技术适用参数[34]
基于以上研究基础,MPE技术已广泛应用于国内外实际的地下水修复工程中. 就目前而言,国外的MPE技术应用发展已比较成熟. 早在1992年,MPE技术便成功应用于美国弗吉尼亚州军需供应中心氯代有机物染污的治理,经过320 d的修复工程,地下水中TCE与PCE的去除率均在98%以上[34]. 我国对于MPE技术的研究虽然相对落后,但处在不断进步之中. 我国利用MPE技术对受二氯乙烯(DCE)、PCE污染的上海一工业仓库地下水进行了治理,成功将目标污染物浓度降至人体健康风险标准以下[35].
虽然MPE技术具有修复效率高、对地面环境扰动较小、可同时对气相、自由相、残留相与溶解相的污染物进行去除等优点[36],但该技术仍存在一定的局限性与缺点. 局限性主要体现在其有一定的适用参数,超出适用范围,很难达到预期修复效果;缺点则主要体现在设备搭建与处理工艺的复杂性上.
2.3 溶解相氯代烃的修复技术
2.3.1原位化学氧化(ISCO)技术
溶解相氯代烃的迁移过程主要受对流与弥散作用的影响,其所造成的污染区域处在动态变化之中,而原位化学氧化技术则主要是通过向地下水中注入一定量的氧化剂来进行氧化修复,氧化剂的迁移同样受对流与弥散作用的控制,因而对处在不断迁移过程中的溶解相氯代烃有较好的修复效果. ISCO技术原理示意如图3所示. 该技术常用的氧化剂主要有臭氧(O3)、高锰酸钾(KMnO4)、Fenton试剂及过硫酸钠(Na2S2O8).

图3 ISCO技术原理示意[37]
O3(氧化还原电位E0=2.07 V)作为一种气态的强氧化剂,主要是通过直接氧化和间接氧化2种方式实现对氯代烃的降解. 直接氧化就是通过O3与氯代烯烃的加成反应实现[38],此氧化方式的氧化效果与O3自身浓度有关,O3浓度越高,自分解率越低,其氧化效果就越好;间接氧化是利用反应产生的具有强氧化性的·OH(E0为2.70~2.80 V)来氧化氯代烃[39],参与反应的O3量越多,生成·OH就越多,降解效果也就越好. O3在pH为5~8的地下水环境中可以发挥良好的降解效果,但生产成本高限制了其在原位化学氧化修复中的应用.
KMnO4(E0=1.68 V)价格低廉,在水中的溶解度较高,是原位化学氧化修复常用的氧化剂. KMnO4对地下水中的TCE、PCE等氯代烃都有比较好的降解效果,可以将其氧化成氯化物和CO2[40]. KMnO4相比于O3在地下水环境中存在的时间更长,适用的pH范围也更广,能够在pH为7~8的地下水环境中实现更好的氧化降解效果. 但KMnO4氧化修复过程中,会有副产物MnO2沉淀产生,对含水层介质造成堵塞,影响KMnO4与污染物的接触,降低修复效果. 针对可以有效吸附MnO2沉淀的蒙脱石等负载材料的研究,是提高KMnO4氧化修复效率的手段之一[41].
Fenton试剂是H2O2和Fe2+的混合试剂,其主要是利用反应生成的·OH来降解氯代烃有机污染物,达到修复目的. 虽然Fenton试剂对氯代烃有较好的修复效果,但其主要适用于pH为2~3的酸性条件,pH过高Fe2+会发生沉淀;另外,H2O2的稳定性差,易分解,在地下水环境中存在的时间极短,这些因素均限制了其在实际污染场地地下水修复中的应用[42]. 目前Fenton试剂的研究方向主要包括环境友好型H2O2稳定剂与改性Fenton试剂的研究.
Na2S2O8是近年来广大学者重点研究的原位化学氧化修复试剂,它能够在一定活化条件下产生具有强氧化性的SO4-·用于降解氯代烃污染物. Na2S2O8相比于其他几种修复药剂稳定性更强,而且适用的pH范围也最广,在pH为2~11范围内均有较好的降解效果,碱性条件下则主要是通过产生·OH降解污染物. 学者们对于Na2S2O8的研究主要体现在对其活化条件的研究,Na2S2O8常用的几种活化方式包括热活化[43]、碱活化[44]、紫外活化[45]、过渡金属离子活化[46]等. 近年来,由于天然含铁矿物储量丰富且价格低廉,有学者利用此类矿物活化Na2S2O8也取得了较好的活化效果[47-49]. 不过大量使用Na2S2O8可能会造成地下水中SO42-增多,这也是在实际修复过程中需要注意的地方.
在实际的地下水修复工程中,除因氧化剂性质不同造成的修复效果差异外,原位注入氧化剂的方式有其固有的缺陷. Chang等[50]利用原位注入过硫酸盐的方式对中国台湾南部工业园区受TCE污染的地下水进行治理时发现,过硫酸盐仅能存在14 d,这与其在未受污染含水层材料中的半衰期差距较大[51]. 出现这种现象的原因在于地下水中有机质与还原性物质对氧化剂的非目标性消耗,限制了氧化剂与目标污染物的有效接触[52]. 当前的许多修复工程是通过加大氧化剂的投加量来提高其对污染物修复效果,但这种方式对于地下水中的微生物多样性以及氧化还原环境造成了极大破坏. 目前,已有学者通过氧化剂缓释材料的研究对氧化剂利用率低的情况作出了改善,提高了氧化剂的长期修复效果[53-55]. 因此研究环境友好型化学氧化剂的缓释材料无论是从成本上还是长期修复效果上都是一个很好的选择.
2.3.2可渗透反应墙(PRB)技术
PRB技术作为一种常用的地下水污染原位修复技术,其主要手段是在受污染地下水下游含水层中,沿地下水流的垂直方向构筑填充有反应材料的可渗透的墙体,受污染的地下水在水力梯度作用下通过PRB 时会与墙体内的填充材料发生物理、化学反应,使污染物得以去除,从而达到修复目的[56]. 由于PRB技术严重依赖地下水流运动,因此对于水溶相氯代烃的治理效果较好. PRB技术原理示意如图4所示.
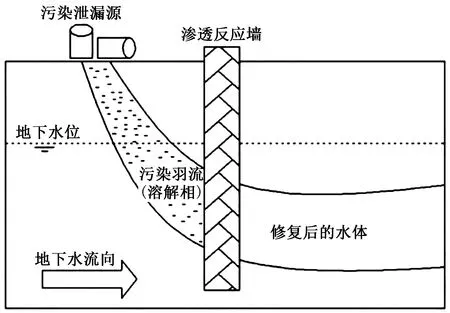
图4 PRB技术原理示意[37]
PRB技术用于修复氯代烃污染的研究进程十分漫长,最早可追溯至1992年,Gillham等[57]首次提出了用零价铁(ZVI)来修复地下水中的氯代烃污染,为PRB填充材料的研究奠定了基础. 1995年1月,在美国加利福尼亚州Sunnyvale首次出现了以ZVI-PRB商业化修复地下水氯代烃污染的案例,开创了PRB商业化修复氯代烃污染的先河[58]. 之后,不少学者研究发现,ZVI比表面积、pH、温度、地下水离子组分、共存有机污染物、表面活性剂及腐殖质等因素均会影响ZVI-PRB修复氯代烃的实际效果,且ZVI比表面积在这些因素中起主要作用[59]. ZVI比表面积越大,其脱氯速率也越快[60]. 因此,具有更大比表面积的NZVI引起了广大学者的关注. NZVI相较于ZVI能够实现对氯代烃污染物更好的降解效果,但其本身也存在易钝化、易团聚的缺点,同时高昂的生产成本也使其在实际污染场地氯代烃的修复上受到限制.
随着PRB技术的不断发展,PRB内的填充材料也呈现出多样性. 有学者利用一些成本较低、天然存在的含铁矿石作为填充材料,实现了对地下水氯代烃良好的降解效果同时去除了一部分重金属污染物与其他有机污染物[61]. 纳米双金属材料也是目前PRB填充材料的一种发展方向. 有研究对合成的纳米铁镍材料展开研究,发现该材料可以增强NZVI的活性并提高对氯代烃的降解效果[62-63]. 也有一部分研究者在此基础上进行负载材料的研究,发现膨润土、壳聚糖都是优秀的负载材料[64-65]. 以实际污染场地污染物性质为考量的PRB填充材料也是目前比较热门的研究方向,Zhou等[66]将活性炭、沸石和ZVI以4∶1∶5的比例混合作为PRB的填充材料,去除了垃圾渗滤液中大部分的COD、总氮、铅镍重金属及多环芳烃.
虽然利用PRB技术处理地下水中氯代烃污染具有成本低、处理效果好等优点,但也存在一定不足,一方面是PRB技术所固有的墙体堵塞问题;另一方面是修复过程中pH与Eh的变化对原本的地下水环境条件造成了破坏[67-68].
2.4 残留相氯代烃的修复技术
2.4.1原位生物修复(ISB)技术
残留相氯代烃大都赋存在含水层空隙介质或者裂隙介质中,在吸附作用和毛细作用下无法迁移,因此其具体状态受含水层环境影响颇深. 而ISB技术主要是利用含水层中特定微生物的新陈代谢活动将有机污染物转化或降解为无污染物质,因此可以有效去除含水层中的残留相氯代烃.
原位生物修复技术对于氯代烃的降解作用可以分为三类:厌氧还原[69]、好氧共代谢[70]和异养同化[71]. 表3为这三类降解作用的特点.
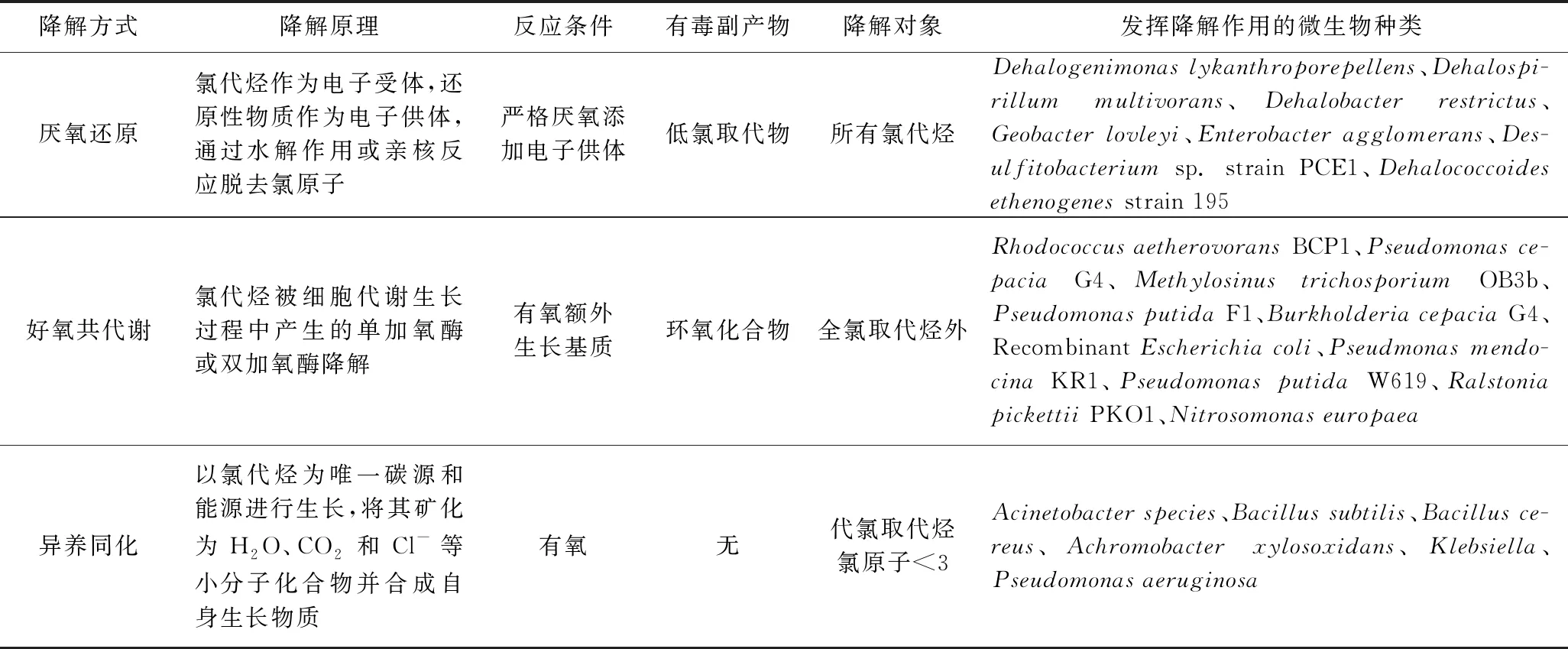
表3 三类降解作用的特点[72-73]
上述三类降解作用特点的研究为实际的生物修复工程指明了方向. 目前,已经有许多污染场地利用原位生物修复技术修复成功的案例,其中较为经典的则是美国爱达荷州受氯乙烯污染地下水的修复案例[74]. 此工程为目前已知世界上规模最大的原位生物修复工程,在向场地周期性注入高浓度的乳酸钠电子供体一段时间后,TCE污染物在生物降解作用下被完全转化,生成了大量无害乙烯. 该案例的经典之处不仅在于实现了对大规模污染场地的成功修复,更重要的是从修复方案的选择到完成修复的整个流程,对于国内复杂地下水污染场地修复工程的设计具有较高的参考价值. 流程主要涉及以下几个方面: ①通过野外中试试验等方式对场地的可生化性进行评估; ②通过抽水试验、示踪试验以及水样分析等方式获得含水层特征参数、污染物组分、污染源位置分布等信息; ③构建场地概念模型,并结合上述已知信息,进行电子供体设计与修复设施的搭建; ④进行修复效果的长期、连续监测,监测指标包含地下水水质参数、污染物及相关降解产物、微生物种类及数目变化、氧化还原参数以及电子供体与营养物质的实际分布等信息[74].
虽然原位生物修复技术具有环境友好与成本低的优点,目前也有许多成功的修复案例,但在对环境条件复杂的实际污染场地修复过程中仍存在一些问题: ①在利用微生物厌氧还原作用对地下水埋深较深的污染场地进行修复时,往往存在菌群数量与活性较低的情况,且还原脱氯的速率会随氯原子数量的减少而剧烈下降,从而造成低氯取代物的大量堆积,达不到污染场地的修复目标. ②在利用微生物的好氧共代谢作用进行污染场地的修复时,添加的生长基质可能会对场地造成二次污染; 另外共代谢作用产生的环氧化合物对微生物有一定的毒害作用,也会造成微生物自身活性的降低,长此以往,可能会出现场地污染修复停滞的问题. ③实际污染场地中能够实现对氯代烃异养同化的微生物种类较少,且其只能够对一些低氯取代物进行降解修复,这些都极大地限制了其在现实生活中的应用.
2.4.2表面活性剂强化抽出处理(SEAR)技术
利用ISB技术修复含水层残留相氯代烃污染的效率较低,有时不能满足实际污染场地治理的要求. 而向地下含水层注入表面活性剂可以增强吸附于多孔介质颗粒的污染物的溶解性能,使含水层介质中的残留相氯代烃逐渐转化为溶解相,可以有效提高抽出处理的效率,因此引起了人们广泛关注.
有学者用室内砂箱模拟试验对SEAR技术与传统抽出处理技术进行了对比,发现SEAR技术可以有效提高对氯代烃的修复效率[75-78]. 效率的提高主要是由于表面活性剂对于污染物的增溶与增流作用[79-80],而这种作用易受污染物性质、含水层介质性质[81]、抽出-回注总量[82]等因素的影响. 同为DNAPL,密度小且溶解度高的氯代烃更容易被抽出去除;含水层介质的性质则主要影响表面活性剂与污染物的接触面积,非均质含水层尤其是低渗透含水层,相较于均质含水层,表面活性剂与污染物的接触受到影响,限制了残留相向溶解相变化的传质过程;抽出-回注的冲刷作用则会产生一种水平驱动力,污染物得到了再迁移与再分布,最大横向迁移距离与污染羽面积都有一定程度的增加,使得表面活性剂与氯代烃的接触面积增大,从而促进了残留相向溶解相的传质过程[83]. 目前,关于SEAR技术修复地下含水层氯代烃污染的研究大多处于实验室规模,可供参考的场地修复工程案例还比较少. 为将SEAR技术成功应用到实际场地修复中,仍存在许多问题需要解决. 首先,针对氯代烃这种DNAPL污染物,表面活性剂的使用会使其界面张力降低,进而促使其发生垂向迁移,增加了更深含水层受污染的风险[84];其次,对含水层中残留表面活性剂的降解产物及其毒理分析的研究还不够完善,不能保证其不会对环境造成影响;最后,针对水文地质条件比较复杂的场地,如何确定表面活性剂的最佳投加量(保证修复达标情况下表面活性剂的最小用量)也是需要考虑的问题. 总之,将表面活性剂用于实际工程的修复仍需要一定规模中试试验数据的支撑;此外,针对复杂水文地质条件污染场地的数值模拟研究也是必不可少的.
2.5 低浓度氯代烃的修复技术
当污染场地氯代烃的浓度较低时,或经过其他修复技术的修复使污染场地氯代烃的浓度降至一定水平时,通常以监测自然衰减(MNA)技术作为污染场地的修复手段. MNA技术主要是在一定监控策略下,利用地下水环境自然发生的物理、化学和生物作用,使得地下水中污染物的浓度在预期时间范围内降至风险可接受水平,从而实现修复目的[85].
针对氯代烃,其自然衰减的途径主要包括两类:一类是以分散、稀释、挥发、吸附等行为为主的非破坏性物理过程;一类是以氧化还原反应为主的非可逆性的化学与生物脱氯过程[86-87]. 其中,针对地下含水层,生物降解过程被认为是氯代烃自然衰减进程中最重要的一个环节[88]. 广大学者在氯代烃自然衰减的室内模拟试验中,不但明晰了好氧氧化与厌氧还原这2种途径下氯代烃的迁移转化机理,而且确立了厌氧还原作用在生物脱氯过程中的主体地位[89-91]. 然而,氯代烃的生物降解过程极易受污染场地水文地质条件的影响,包括污染场地的氧化还原环境与存在的特征离子等因素[92]. 例如,硫酸盐和硫化物可以抑制硫酸盐还原菌的脱氯作用[93-94]. 此外,主要的脱氯细菌,如Desulfuromonassp.[95]和Dehalobactersp.[96],只能将PCE脱氯为顺式二氯乙烯(cis-DCE),Dehalococcoidessp.[97]是唯一可以将PCE转化为无害乙烯(ETH)的细菌,但也存在cis-DCE到氯乙烯(VC)以及VC到ETH脱氯效率低的问题,因此有时会在地下水中残留有毒的中间体,如cis-DCE和VC. 因此,在利用MNA技术进行修复前要对场地的自然衰减能力作出评价. 一方面,需要收集场地历史的地下水监测数据,明晰污染物浓度随时间的变化关系,进而判断自然衰减能否控制污染范围的扩大与浓度的降低;另一方面,需要借助地下水的一些基本理化性质(如pH、温度、总有机碳、电导率、溶解氧等)以及电子受体(如O2、NO3-、SO42-、Fe3+)含量的变化进行环境水文地球化学指标的评估,间接判断生物降解作用的发生[98];同时,也可以利用微宇宙试验模拟污染场地实际的水文地质条件[99],直接验证自然衰减的发生,并结合稳定同位素示踪技术进一步了解污染物降解的中间产物以及实际的生物降解路径[100]. 目前,已有学者利用MNA技术成功指导实际场地的污染修复工作. Larsen等[101]利用当地的树芯样品对捷克共和国北部一工厂的污染情况进行了监测,监测结果显示该工厂地下水主要受PCE、TCE、cis-DCE这3种氯代烃的污染,其浓度分别可达230、14和72 mg/L;历经8年的自然衰减,总氯乙烯(CE)>1 mg/L的污染羽尺寸由8 hm2减至1 hm2. Kawabe等[92]对日本高富市受污染地下水场地的自然衰减状况进行了监测,监测结果显示,挥发性有机物(VOC)的风险水平远低于刚发现时,并且在2033年之前可以实现当地VOC污染的完全修复;但是,在监测过程中,PCE、TCE、cis-DCE的风险水平有时会出乎意料地增加.
上述两经典的实际修复案例为未来MNA技术的应用提供了极大的指导意义. 一方面,在地下水污染物浓度的监测时,要充分调研当地的水文地质条件,因地制宜地选择适当的指标来反映污染物浓度的变化,在不影响监测数据准确性的同时可以最大程度地降低监测成本;另一方面,为实现场地污染物的完全修复,需进行全面、连续的长期监测,避免个别位点修复未达标状况的发生,同时这也对场地的监测条件以及环境管理提出了更高的要求.
3 结论与展望
a) 地下水氯代烃主要有气相、自由相、溶解相、残留相4种赋存状态,它们在一定条件下可以相互转化,这极大地增加了污染场地的治理难度.
b) 针对氯代烃不同赋存状态,其适用的修复技术也存在差异性. 其中,气相抽提(SVE)技术与热处理强化(TE)技术适用于气相氯代烃修复;多相抽提(MPE)技术适用于自由相氯代烃修复;原位化学氧化(ISCO)技术与可渗透反应墙(PRB)技术适用于溶解相氯代烃修复;原位生物修复(ISB)技术与表面活性剂强化抽出处理(SEAR)技术适用于残留相氯代烃修复;监控自然衰减(MNA)技术则可以用于低浓度氯代烃的长期修复或与其他技术的联用.
c) 根据当前修复技术的发展现状,PRB技术墙体新介质的开发、缓释氧化剂及污染物靶向修复材料的开发、高抗性且生存能力强的工程菌的培育以及修复技术之间的联用将是氯代烃污染治理的发展方向;另外,在修复技术的选择与联用上,必须遵循技术与场地水文地质条件相适应的修复思路.
