HPLC-MS/MS测定煎炸植物油中羧甲基赖氨酸和羧乙基赖氨酸
2021-07-13江小明周玮婧涂凤琴
杨 明,江小明,王 澍,何 平,侯 靖,杨 永,邵 梅,周玮婧,涂凤琴
武汉食品化妆品检验所,湖北 武汉 430040
晚期糖基化终末产物(AGEs)是由还原糖及其氧化产物的羰基与蛋白质、氨基酸、脂类或核酸的游离氨基经过缩合、重排、裂解及氧化修饰等一系列反应产生的一类复杂化合物的总称[1]。AGEs主要包括羧甲基赖氨酸(CML)、吡咯啉(Pyr)、3-脱氧葡萄糖酮酸(3-DG)、羧乙基赖氨酸(CEL)等,其中对CML和CEL的研究最多,CML通常被视为食品中AGEs产生的主要标志性物质。研究表明,人们从食物中摄入的AGEs在体内不断累积,与糖尿病、衰老和动脉粥样硬化等疾病的发生发展有密切关系[2-6]。食用植物油常被用来煎炸畜禽肉、果蔬和面制品等,有研究报道油脂在高温煎炸过程中可能会促进食品中AGEs的形成,其通过发生过氧化产生二羰基化合物,进而与蛋白质的氨基交联形成AGEs[7]。目前AGEs的研究对象主要集中在畜禽肉、蛋、乳制品等食品基质,而关于煎炸植物油中AGEs的研究及检测方法报道相对较少。因此,建立煎炸植物油中AGEs的检测方法,对于进一步探索油脂参与美拉德反应形成AGEs的机制研究具有重要意义。
目前,食品中AGEs的检测方法主要有气相色谱-质谱联用法(GC-MS)[8-9]、酶联免疫法(ELISA)[10]、高效液相色谱法(HPLC)[11-12]和液相色谱-串联质谱法(LC-MS/MS)[13-15]等。ELISA法操作简便快速,但定量分析结果准确性较差。HPLC法和GC-MS法灵敏度较高、分析准确性高。除CML和CEL外,多数AGEs具有自发荧光,可直接采用高效液相色谱-荧光检测法(HPLC-FLD)进行测定。HPLC-FLD或GC-MS法测定CML和CEL时,需将样品进行衍生化处理,操作步骤较为烦琐。而HPLC-MS/MS不仅具有高选择性、高灵敏度和优异的定性定量能力等特性,而且无须进行复杂的样品衍生化处理就可实现直接测定CML和CEL的目的。目前,已有较多文献报道利用HPLC -MS/MS测定油条、液态牛奶、奶粉、饼干、面包、酱油和食用油煎炸食物等食品基质中CML和CEL含量[16-18]。采用HPLC-MS/MS法测定煎炸植物油中AGEs检测方法鲜见报道。
作者利用HPLC-MS/MS建立了一种测定煎炸植物油中CML和CEL的分析方法。样品提取液经MCX固相萃取小柱萃取净化,采用NH2色谱柱进行分离,前处理过程无须进行复杂的柱前衍生,操作步骤相对简单,分析时间短,重现性好。
1 材料与方法
1.1 仪器和试剂
Agilent 1260高效液相色谱仪、Agilent 6460质谱仪、固相萃取装置:USA Agilent公司; Vortex-Genie 2涡旋振荡器:USA SCND公司;乙腈、甲酸、甲醇、氨水、二氯甲烷:HPLC 级,GER Merck KgaA 集团;羧甲基赖氨酸(C8H16N2O4, 纯度98%)、氘代羧甲基赖氨酸(CML-D4, C8H12D4N2O4, 纯度98%)、羧乙基赖氨酸(C9H18N2O4, 纯度98.6%)和氘代羧乙基赖氨酸(CEL-D4, C9H14D4N2O4, 纯度91.1%)均为First Standard标准品;去离子水(18.2 MΩ·cm)由Milli-Q纯水仪制得;HLB固相萃取柱(60 mg/3 mL): USA Waters公司;MCX固相萃取柱(60 mg/3 mL,活化液:甲醇、水各3 mL):博纳生物公司。
1.2 标准溶液配制
准确称取一定量的CML、CEL标准品于25 mL容量瓶中,用乙腈-水(体积比为90∶ 10)溶解定容至质量浓度为10 μg/mL,于-18 ℃下储存。临用时以空白样品基质提取液配制成系列质量浓度的标准工作溶液,用于制作标准曲线。
1.3 样品处理
1.3.1 提取
称取1.0 g煎炸植物油试样于50 mL塑料称量管中,加入适量内标和2 mL二氯甲烷,混均,加入10 mL 0.1%甲酸水溶液,振荡提取10 min后离心4 min(转速3 950 r/min),收集上清液,再重复提取1次,合并2次提取液并定容至20 mL,待用。
1.3.2 净化
准确移取10 mL提取液过已活化MCX固相萃取柱,弃去滤液,再依次用水、甲醇各3 mL淋洗,让淋洗液流出,抽干小柱,用5 mL 5%氨水甲醇洗脱,洗脱液经氮吹浓缩后,用1 mL乙腈-水(体积比为90∶ 10)复溶混匀,过膜后待测。
1.4 试验条件
1.4.1 色谱条件
Shodex Asahipak NH2P-50 2D色谱柱(规格150 mm×2.0 mm, 5 μm);进样量2 μL;柱温40 ℃;流动相A为含0.1% HCOOH的水溶液,流动相B为乙腈;梯度程序:0~1.5 min,90% B;1.5~5.0 min,90%~30% B;5.0~7.0 min,30% B;7.0~7.7 min,30%~90% B;7.7~11.0 min,90% B;流速0.005 mL/s。
1.4.2 质谱条件
离子源,AJS ESI源,正离子;毛细管电压(IS)4 000 V;干燥气温度250 ℃;鞘气温度320 ℃;鞘气流速11 L/min;干燥气流速5 L/min;碰撞气体为氮气;多反应监测(MRM)模式。
2 结果与分析
2.1 色谱条件优化
比较了Waters XBridge C18色谱柱(150 mm×2.1 mm, 5 μm)、ACQUITY UPLC BEH HILIC色谱柱(100 mm×2.1 mm, 1.7 μm)和Shodex Asahipak NH2P-50 2D氨基色谱柱(150 mm×2.0 mm, 5 μm)等不同填料的色谱柱对CML和CEL的分离效果,结果见图1。由于CML和CEL具有强极性,在C18柱上保留很弱,出峰时间快。待测物与杂质很难分离,在HILIC柱中保留效果有所改善,但CML与CEL的分离度不够理想。而NH2柱不仅能较好地保留CML和CEL,而且可实现待测物的有效分离。因此选用Shodex Asahipak NH2P-50 2D氨基色谱柱进行下一步试验分析。
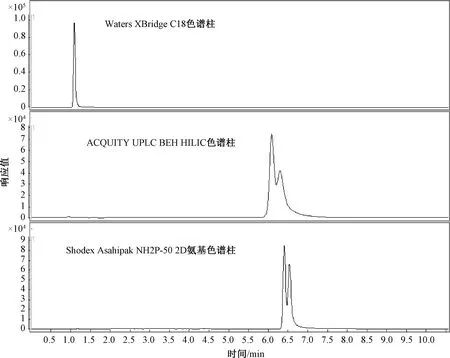
图1 不同填料的色谱柱测定CML和CEL的色谱图Fig.1 Chromatograms of CML and CEL determined by different packed columns
比较了乙腈-水、甲醇-水等流动相体系对CML、CEL、CML-D4和CEL-D4的分离效果。结果表明目标物在乙腈-水组成的流动相中响应较高,因此选择乙腈-水为色谱流动相。在流动相中加入甲酸可以提高正离子模式下物质的离子化效率,进而增强其质谱响应。试验表明,在水相中加入0.1%甲酸(HCOOH)时,各目标物的峰响应显著提高。进一步考察了不同比例的初始流动相(70%乙腈-30%水、80%乙腈-20%水、90%乙腈-10%水、95%乙腈-5%水)对各目标物的保留时间和峰形的影响,结果表明以90%乙腈-10%水(含0.1% HCOOH)为初始流动相时,目标物的分离效果最佳,响应高且峰形对称。为增强目标物的分离效果,试验经优化采取梯度洗脱程序。
2.2 提取溶液的选择
由于CML、CEL极性强,易溶于水、甲醇、乙腈等极性溶剂,故在样品提取过程中以水、甲醇-水和乙腈-水为提取溶剂,考察不同溶剂比例(10%~100%)下的提取效率,结果发现以高比例的水为提取溶剂时效果较好,100%水提取效果最佳。进一步比较添加不同含量甲酸的水溶液作为提取溶剂时的提取效率,结果表明酸度对提取效果的影响不显著。考虑后续净化,选择0.1%甲酸水溶液作为提取溶剂。
2.3 固相萃取净化小柱的选择
考察了MCX复合型阳离子交换柱、HLB固相萃取柱及C18固相萃取小柱对提取液的净化效果。CML、CEL极性强,在C18固相萃取小柱上完全无保留,随上样液一起流出。HLB固相萃取柱对CML和CEL有一定的保留作用,但净化回收率低,均低于60%。MCX复合型阳离子交换小柱对提取液的净化效果最佳,保留效果好,CML和CEL的回收率高,且回收率均高于90%。因此,采用MCX复合型阳离子交换小柱净化。
2.4 质谱条件优化
分别配制质量浓度为500 ng/mL的CML、CML-D4、CEL和CEL-D4标准溶液,以10 μL/min 的针泵流速在不同模式下进行母离子全扫描,结果表明,在电喷雾正离子扫描模式下,各化合物的质谱响应良好,灵敏度高。在一级质谱扫描模式下,目标物均产生 [M+H]+的母离子。再根据二级质谱扫描得到的二级碎片离子质谱图,选取响应良好、干扰少的两对子离子组成监测离子对,并进一步优化每个离子对的最佳碰撞能量(CE),使得每种化合物的离子化效率达到最佳。CML、CML-D4、CEL和CEL-D4的质谱参数列于表1。图2为 CML、CML-D4、CEL和CEL-D4标准溶液的多反应监测(MRM)色谱图。

表1 CML、CML-D4、CEL和CEL-D4的质谱参数Table 1 MS parameters of CML, CML-D4, CEL, and CEL-D4

图2 CML、CML-D4、CEL和CEL-D4标准溶液的多反应监测(MRM)色谱图Fig.2 Chromatograms of MRM for CML, CML-D4, CEL, and CEL-D4 standard solutions
2.5 基质效应
基质效应(ME)是指提取液中的非目标物组分对目标物的离子化分析引起的增强或抑制效应[19-21]。ME通常以基质标准曲线斜率(K1)与溶剂标准曲线斜率(K2)的比值来表示,ME在0.85~1.15时,基质效应较弱;ME大于1.15时,存在较强的基质增强效应;ME小于0.85时,存在较强的基质抑制效应。试验采用空白样提取液和纯溶剂分别配制同一标曲点进行基质效应的评价。结果表明,实际样品基质对CML、CEL存在一定的离子化抑制效应,ME分别为0.83和 0.87。采用内标法定量时,CML、CEL的基质效应不明显,基质效应对测定结果的影响得以补偿。因此,本方法在样品中添加同位素内标,采用内标法空白基质标准工作曲线定量,使定量结果更加准确可靠。
2.6 线性关系、检出限与定量限
以内标法配制空白基质标准工作溶液,采用本方法进行测定,以外内标峰面积比为纵坐标(y)、对应外内标质量浓度比为横坐标(x)绘制标准曲线。结果表明,CML、CEL在2.5~100 ng/mL范围内线性关系良好,CML线性方程为y=1.31x-0.060 5,r=0.999 5;CEL线性方程为y=1.66x-0.007 8,r=0.999 5。将标准溶液添加到阴性样品中,在优化的前处理和测定条件下,CML和CEL的LOD、LOQ分别均为2.0 μg/kg、5.0 μg/kg。
2.7 回收率和精密度
分别在煎炸植物油空白基质样品中添加3个不同质量浓度水平(5.0、10.0和100.0 μg/kg)的标准溶液,按该方法预处理,每个添加水平测定6次,计算回收率和精密度。CML平均回收率为91.8%~105.4%,相对标准偏差(RSD,n=6)为2.8%~7.8%。CEL平均回收率为97.8%~102.1%,RSD为2.8%~6.5%(表2)。结果表明,该方法回收率高、稳定性好,适用于煎炸植物油中CML和CEL的测定。

表2 CML和CEL的平均加标回收率和相对标准偏差(n=6)Table 2 Average spiked recovery and the relative standard deviations of CML and CEL(n=6)
2.8 实际样品测定
对不同温度(100~180 ℃)、不同时间(1~36 h)条件下煎炸面窝后的植物油进行测定,固定煎炸时间为30 min,每升高20 ℃取样1次。在180 ℃下,每1 h取样1次。结果表明,在不同煎炸时间、不同煎炸温度下的煎炸植物油样本中均未检出CML和CEL。
3 结论
建立了煎炸植物油中CML和CEL的高效液相色谱-串联质谱(HPLC-MS/MS)检测方法。该方法采用0.1%甲酸水为提取溶剂,样品提取液经MCX混合型阳离子交换固相萃取小柱净化,采用氨基色谱柱进行分离,前处理无须进行柱前衍生,过程简便、快捷、高效。采用同位素内标法空白基质标曲校正,提高了定量结果的准确性和可靠性,适用于煎炸植物油中CML和CEL的快速检测。
