金催化炔基苯并二英环化合成8-羟基异香豆素的理论研究
2021-07-11杨一莹朱荣秀张冬菊刘成卜
杨一莹,朱荣秀,张冬菊,刘成卜
(山东大学化学与化工学院,理论化学研究所,济南250100)
异香豆素是苯并吡喃酮类化合物的总称,普遍存在于天然产物、药物分子及有机合成中间体当中[1,2],具有多种药理作用[3~5].该类化合物最具代表性的是8-羟基异香豆素,由于其8号位羟基的较高活性,在抗真菌和抑制组胺活性物质释放等方面有广泛应用[6,7].因此,8-羟基异香豆素的高效精准合成是备受关注的热点课题.
8-羟基异香豆素的合成一般是通过邻炔基苯甲酸(酯)或苯甲酰胺的环化予以实现[8~10],反应条件通常较为苛刻,并且涉及多个反应步骤,致使产物收率较低[11,12].开发条件温和、简单高效、普适性广、选择性高的新型合成方法一直是具有挑战性的课题.2017年,Mohapatra课题组[13]报道了金催化炔基苯并二英环化高效合成8-羟基异香豆素的方法,并提出了相关的反应机理(Scheme 1).他们发现,三苯基膦六氟锑酸金络合物[(Ph3P)AuSbF6]可高效率催化该反应,异香豆素产率可达85%.他们认为反应涉及底物配位、羰基亲核环化、水分子亲核加成、丙酮消除、质子化脱金等过程,炔基苯并二英(R)经历金-π络合物(A)、烯基金中间体(B)、烯基金-H2O加合物(C)、产物前驱物(D)等衍变为目标产物(P).该反应机理的可行性需要用理论化学方法予以验证.本文通过密度泛函理论(DFT)研究了Scheme 1所示的典型反应,给出了新的反应机理,期望对相关的实验研究提供一定的理论指导.

Scheme 1 Au(I)-Catalyzed cycloisomerization of phenylethynyl-benzodioxin(R)synthesizing 8-hydroxy-3-substituted isocoumarin(P)[13]
1 计算方法
所有计算使用Gaussian 09程序[14]完成.结构优化均应用极化连续介质模型(PCM)[15,16],考虑了溶剂化效应,使用的溶剂为四氢呋喃,与实验一致.计算采用了能较准确描述弱相互作用的M06泛函[17,18],对于Au,Ag和Sb原子,选用LanL2DZ[19]赝势基组,并增加了f-型[20]极化函数,其轨道指数分别为1.050,1.611和0.218;对体系中的其它非金属原子,采用6-31G(d,p)[21]标准基组.由于计算体系较大,对金催化剂中的3个苯基进行了简化,使用甲基代替,已有研究表明,这种非活性中心位置的简化对结果影响较小[22].为验证简化模型的合理性,对决定反应速率的中间体和过渡态使用真实催化剂进行相关计算.已通过频率计算确认了全部稳定点的性质,确保局域最小点无虚频,过渡态(一级鞍点)有且仅有一个虚频.为了识别最低能量路径,对全部一级鞍点进行了内禀坐标路径解析(IRC)[23],确认了每个过渡态连接的反应物和产物.自然轨道(NBO)布局分析[24]使用Gaussian 09软件包中的NBO 3.1程序[25]完成,一些关键结构的三维(3D)图采用CYLview可视化软件[26]构建.
2 结果与讨论
2.1 底物配位

Fig.1 Chemical structures of complexes of the model catalyst(Me)3PAuSbF6 with phenylethynyl-benzodioxin(R)with calculated bond distances(nm)and relative Gibbs energies(kJ/mol,in parentheses)
2.2 羰基亲核环化

Fig.2 Calculated Gibbs energy profiles with schematic structures for the Au-catalyzed 5-exodig and 6-endo-dig cyclizations of phenylethynyl-benzodioxin(R)

Table 1 Calculated distortion/interaction energies for regioselectivity-determining transition states
2.3 H2O分子亲核加成
Mohapatra等[13]认为,烯基金中间体一旦形成,将与H2O形成分子间加合物,继而H2O分子加成与酮分子消除协同进行,导致第2个烯基金中间体的生成(Scheme 1).我们尝试模拟这样的反应过程,但经多次计算,无法证实此实验的猜测.因此我们认为,不存在Scheme 1中建议的H2O分子亲核加成机制.计算给出了与实验猜测明显不同的结果,一方面,亲核加成并不涉及C(sp2)—O键的断裂(如Scheme 1中结构C所示),而是导致C(sp3)—O键的断裂;另一方面,H2O分子亲核加成与酮分子消除不是协同发生而是按照先后顺序进行.
图3给出H2O分子亲核加成的4种机理(路径Ⅰ~Ⅳ).路径Ⅰ和Ⅱ基于Mohapatra等[13]的猜测,H2O分子中带负电荷的O亲核进攻C1(sp2)原子,但在两个路径中H2O分子中H原子的迁移方向不同,在路径Ⅰ中迁移到O1(TS3),能垒为221.6 kJ/mol,导致C1(sp2)—O1键断裂;而在路径Ⅱ中迁移到O2(TS4),能垒为202.6 kJ/mol,导致C2(sp3)—O2键断裂.由于p-π共轭效应,C(sp2)—O键(键能320.5 kJ/mol)[29]比C(sp3)—O键(键能275.0 kJ/mol)更强[30],在反应中更难断裂,与通过NBO分析计算的键级一致[图3(A)中IM3,两个C(sp3)—O键键级分别为0.89和0.77,而两个C(sp2)—O键键级分别为1.13和0.98],所以路径Ⅰ的能垒更高.另外,TS3为四元环过渡态,TS4为船式六元环过渡态,均有较高的环张力,导致两条路径均涉及较高的能垒,与温和的反应条件不一致,故可排除这两条路径发生的可能性.
在路径Ⅰ和路径Ⅱ涉及的两个过渡态结构中,O的亲核进攻与H迁移是协同过程,研究表明,体系中的H2O分子通常可以协助这样的过程,为此,将另外一个H2O分子引入活性中心,协助反应的进行,计算结果如图3(B)中路径Ⅲ和Ⅳ所示,分别对应于路径Ⅰ和路径Ⅱ.在这两条路径中,一个H2O分子作为反应物,另一个则扮演质子梭的角色,协助质子迁移.路径Ⅲ涉及椅式六元环过渡态(TS5),能垒为129.0 kJ/mol,导致C1(sp2)—O1键断裂;而路径Ⅳ涉及八元环过渡态(TS6),能垒为135.4 kJ/mol,导致C2(sp3)—O2键断裂.与非水分子协助的路径Ⅰ和Ⅱ相比,能垒明显降低,但与温和的反应条件仍不相符,需要进一步寻找另外可能的反应路径.
上述反应路径中,H2O中O亲核进攻的是C1(sp2)原子,另一种可能性是进攻C2(sp3)原子.因此,进一步考虑了H2O分子在C2(sp3)原子上亲核加成的两种方式:从氧杂苯环一侧进攻(路径Ⅴ)或从苯环侧进攻(路径Ⅵ),计算结果示于图4.在路径Ⅴ中,H2O分子通过TS7对C2(sp3)原子进行亲核进攻,导致C2(sp3)—O2键断裂,能垒高达190.6 kJ/mol,在室温下显然难以发生.有趣的是,当H2O从苯环侧进攻时(路径Ⅵ),如TS8所示,导致C2(sp3)—O1键断裂,反应能垒下降到62.0 kJ/mol.比较发现,TS8虽与TS7有类似的构型,但C2(sp3)原子与H2O分子中O原子之间的距离明显不同,TS8中为0.274,而TS7中为0.235 nm,导致反应按不同的历程进行.路径Ⅴ可以描述为H2O分子加成与C2(sp3)—O2断裂协同发生的过程,而路径Ⅵ是一个分步过程,C2(sp3)—O1断裂(TS8)先于H2O分子加成(TS9).计算结果表明,沿路径Ⅵ,反应需要克服的能垒下降到62.0 kJ/mol,是最有利的亲核加成路径,匹配实验中温和的反应条件.
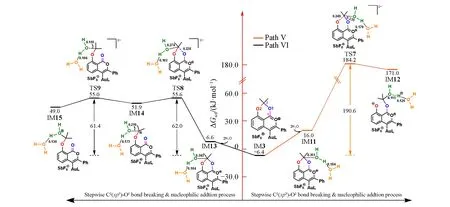
Fig.4 Calculated Gibbs energy profiles with schematic structures for the nucleophilic addition process of water molecular at C2(sp3)site
2.4 丙酮消除和质子化脱金
H2O分子亲核加成导致的中间体IM15是一个两性离子中间体,其中正电荷转移到水分子氧原子上.IM15经丙酮消除和质子化脱金形成目标产物8-羟基-3-苯基异香豆素(P),计算结果示于图5.
由图5可见,中间体IM15经TS10转化为另一个两性离子中间体IM16,该过程是一个质子转移过程,其中水分子作为质子梭传递质子.IM16经C2—O2单键旋转得到更稳定的中间体IM17,同时,酚羟基上的氢转移到环酯单元的羰基氧(O1)上,该异构化过程放热41.0 kJ/mol.IM17经TS11转化为IM18,在TS11中,C2(sp3)—O2键断裂与质子转移过程同时发生,实现丙酮分子的消除,该过程的能垒为33.4 kJ/mol.在IM18中,丙酮与水合质子通过较强的氢键形成“丙酮-水合质子”单元,该单元迁移到金附近,形成质子脱金前驱物IM19.最后,质子脱金过程通过TS12发生,其能垒仅为5.9 kJ/mol,是一个非常容易的过程,得到“产物-金催化剂-水分子-丙酮”4组分络合物IM20.最后,IM20经配体交换得到目标产物、再生IM1,释放水分子和副产物丙酮,开启下一个催化循环.

Fig.5 Calculated Gibbs energy profile with schematic structures for the acetone elimination and protodeauration processes from IM15
另外,计算结果表明,副产物丙酮中的氧来自于反应体系中水的贡献,与实验猜测不同.根据Scheme 1,丙酮中的氧来自于底物.可通过进一步的同位素标记实验来证实丙酮中氧的来源.

Fig.6 Overall Gibbs energy profile for the title reaction
