分子形貌所指示的羟基卡宾及其衍生物的质子转移反应
2021-07-11赵东霞张海霞冯文娟杨忠志
赵东霞,张海霞,冯文娟,杨忠志
(辽宁师范大学化学化工学院,大连116029)
卡宾自20世纪50年代被引入有机化学以来受到研究者广泛关注[1~4].羟基卡宾分子是卡宾中具有研究价值的一类,其主要通过羰基、酰基等直接质子转移得到,很容易分解生成醛,是一种非常有用的合成中间体[5~7].
卡宾分子H2C∶由于具有2个未成对电子,根据电子自旋排列方向不同,分别呈现出单线态和三线态.对于三线态卡宾,中心碳原子呈sp杂化,2个sp杂化轨道分别和2个氢原子成键,2个未杂化的p轨道分别容纳一个电子,因此,三线态卡宾可以看作是一个双游离基.而单线态卡宾的中心碳原子呈sp2杂化,在3个杂化轨道中有2个杂化轨道与氢原子成键,另外一个杂化轨道只容纳2个电子,不参加杂化的轨道是空轨道,其既不与其它原子成键也不容纳电子,这样的电子排布情况尽最大可能地减少了电子之间的相互排斥.Bacskay等[8]运用Gaussian-2探究了衍生物HCNO,HNCO和环N(H)CO在反应过程中单线态与三线态能量的大小,通过对比相同结构的能量,发现在反应过程中单线态的能量比三线态的能量低得多.Hirai等[9]对亚甲基卡宾进行了研究,通过对比发现三线态比单线态稳定.因此,不同结构卡宾分子的单线态和三线态稳定性不同.
羟基卡宾分子(HOCH)质子转移过程中存在一个独特之处,即在反应过程中随着反应进行,羟基中的H原子在对应C—O键的上方来回移动,过渡态处H原子位于C和O原子之间,O原子同时与C原子和H原子相连,形成分子内氢键[10,11].根据大量理论以及实验研究能够得到的结论是,羟基卡宾通过质子转移反应转化为醛,这使羟基卡宾化合物的应用更加广泛[12~19].但是对羟基卡宾衍生物的研究仍然有待深入.
依据电子运动的经典转折方程,杨等建立了分子形貌理论[20].分子的电子密度可决定分子的所有性质,然而在三维空间中却难以表现,分子形貌(Molecular face,MF)不仅能够表现分子的大小和形状,还能够展示分子内禀电子转折界面上的前沿电子密度分布,是同时表征分子的外在和内在性质的量子化学模型.前沿电子密度不是前线分子轨道.目前,使用该方法研究了一些无机、有机和生物小分子的分子形貌[20~25]、H与H原子形成H2过程的分子形貌的动态变化[22],以及H+F形成HF分子过程中H与F原子的相互作用和变化[26]、F+C2H4反应过程中F与乙烯的相互作用及两者界面轮廓的变形情况[27]、SN2反应(C和Si)[28,29]、以及烯烃+卤化氢的马氏和反马氏等反应[30],形象地展示了原子和分子间极化和成键的图像,为描述化学反应机理提供一个直观和形象的工具.
本文使用从头计算方法和分子形貌理论,研究了羟基卡宾及其衍生物HOCX(X=—H,—CN,—NO2,—COOH,—CHO,—COCH3,—F,—Cl,—Br,—CH3,—OH,—OCH3,—NHCH3,—NH2)的质子转移反应,并对其反应所涉及化学键的形成和断裂、分子表面积和体积的变化,以及分子形貌的动态变化进行了研究,进而得出分子形貌指示了上述反应.
1 理论模型和方法
1.1 分子中1个电子所受到的作用势
分子中1个电子所受到的作用势(Potential acting on an electron in a molecule,PAEM)是由杨忠志等[31~34]率先提出并研究的,使用其定义分子的形貌.PAEM为分子中的1个电子受到体系所有原子核以及其余电子的作用势能,表达为
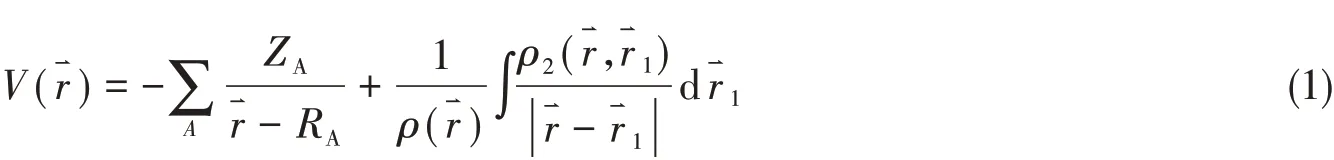
式中:右侧第一项为受到所有原子核的吸引作用势,第二项为受到其余电子的作用势,式中的各个物理量均采用原子单位.其中,ZA为A原子的核电荷数,RA为A原子核的坐标指该电子和其余任何1个电子的坐标是指1个电子出现在的几率密度;指1个电子出现在处、同时另一电子出现在处的双电子几率密度.在组态相互作用下,推导出单电子密度和双电子几率密度的具体表达式[33,34].使用Davidson等[35]发展的MELD程序包,可以计算出单电子密度和双电子密度表达式中的分子积分,使用自编的PAEM程序,可以计算出分子中1个电子出现在⇀r处所受到的作用势
1.2 分子形貌
分子形貌描述的是分子的形和貌[8]:形为分子的形状和大小,貌为前沿边界面上的电子密度,就像分子的身份证,是唯一定义的.分子形貌界面,是电子运动的经典转折点,其内部为电子运动的经典允许区域,而其外部为电子运动的经典禁阻区域,其主要思想为:分子中的1个电子在运动的过程中,其动能和势能均在不断变化,当该电子逐渐远离原子核区域时,其动能不断减小,势能不断升高,当动能减小到零时,该电子能量等于它的势能,此时,该电子所处位置称为电子运动的经典转折点.即,当1个在⇀r处的电子所受到的势能与该分子第一电离能负值相等时,该点就是分子内禀特征轮廓点,这些点的集合就为分子内禀特征轮廓(Molecular intrinsic characteristic contour,MICC),可以表达为

式中:I为分子的第一垂直电离能;⇀r为分子内禀特征轮廓点;G为MICC点的集合;V(⇀r)代表在分子的内禀特征轮廓点处单电子所受到的作用势(PAEM),首先计算出电子在空间各点的V(⇀r)PAEM和分子的第一电离能,然后依据式(2)可以获得MICC,使用Matlab软件,将电子密度附着在MICC上,并以颜色表示,即得到分子形貌MF.
1.3 计算细节
1.3.1 几何结构优化、内禀反应坐标验证和第一垂直电离能 在M06-2X/6-311++G(d,p)理论水平下,
对羟基卡宾分子及其衍生物进行几何结构优化,得到只有一个虚频的过渡态结构,并进行内禀反应坐标(IRC)验证.在相同的理论水平下,计算这些反应的活化能以及所涉及分子的第一电离能.
1.3.2 PAEM和MF的计算 使用Davidson等[17]发展的MELD程序包,在CISD/6-311++G(d,p)理论水平下结合自编程序计算PAEM.首先,在所要研究的分子体系周围,分别沿着x轴、y轴和z轴,每隔固定间距取1个值,建立1个三维的栅格盒子;其次,利用MELD精密从头计算程序并结合自编程序,计算出每个电子坐标位置处该电子所受到的PAEM.然后利用式(2)获得分子的MICC,计算出体积和表面积;最后将分子电子密度映射在该分子的MICC上,使用Matlab程序包绘制出分子形貌MF.
1.4 实 例
图1示出了HCl,CH2==CHCH3和CH2ClCH2COOH的MF,颜色标尺MFED为MF上的电子密度的值,即前沿电子密度,红色到蓝色的颜色变化表示电子密度数值由小到大的渐变值,对于卤化氢与烯烃的加成反应,HCl+CH2CHCH3反应符合马氏规则,反应的主产物为CH3CHClCH3;由于双键C原子上有吸电子基团—COOH,所以HCl+CH2CHCOOH加成反应的方向是反马氏规则的,反应的主产物为CH2ClCH2COOH.HCl分子MF图[图1(A)]中,H原子区域边界面是MFED最小值区域,CH2==CHCH3的MF图[图1(B)]中,π键区域是前沿电子密度大的区域,双键的第一个碳原子上方界面的前沿电子密度(7.097×10-3)大于第二个碳原子的(5.839×10-3),所以H原子加成到第一个碳原子,而Cl原子加成到第二个碳原子,符合马氏规则,CH2==CHCOOH的MF图[图1(C)]中,前沿电子密度大的π键区域,双键的第一个碳原子上方界面的前沿电子密度值(6.147×10-3)小于第二碳原子的(6.978×10-3),所以H原子加成到第二个碳原子,而Cl原子加成到第一个碳原子,符合反马氏规则,使用分子形貌理论可以得出与通常化学认识一致的结果,从上面的描述可以得出,使用分子的内禀电子转折边界面的前沿电子密度,可以指示分子间相互反应的位点和方向[14].

Fig.1 Molecular faces of the HCl(A),CH2=CHCH3(B)and CH2=CHCOOH(C)moleculeds
2 结果与讨论
羟基卡宾同时具有单线态和三线态,质子化反应路径基本相似,图2示出了羟基卡宾分子及其衍生物质子转移反应过程中反应物、过渡态和产物的几何结构示意图.无论是单线态还是三线态下的反应,羟基上的H原子发生转移,与C原子形成C─H化学键.

Fig.2 Structure schemes of proton transfer reactions of hydroxyl carbene and its derivatives HOCX(X=—H,—CN,—NO2,—COOH,—CHO,—COCH3,—F,—Cl,—Br,—CH3,—OH,—OCH3,—NHCH3 and—NH2)
2.1 羟基卡宾分子及其衍生物质子转移反应的反应活化能
在M06-2X/6-311++G(d,p)理论水平下,计算得到单线态和三线态羟基卡宾分子及其衍生物质子转移反应过程中的反应活化能,示于图3,其反应的活化能和过渡态结构的虚频均列在表S1(本文支持信息)中.RC(Reaction complex)代表反应物,TS(Transition state)代表过渡态,PC(Product complex)代表产物,纵坐标为相对能量,横坐标为反应坐标.

Fig.3 Activation energies of the proton transfer reactions for singlet and triplet hydroxyl carbene and their respective derivatives
从图3(A)可见,对于简单的羟基卡宾质子转移反应来说,单线态和三线态下的质子转移反应路径相似,从反应物到过渡态,能量逐渐升高,过渡态处能量最高,从过渡态到产物,能量逐渐下降.由势垒图可见,单线态的羟基卡宾更稳定,反应活化能高于三线态,反应更难发生,这与上面提到的Bacskay等[3]的研究结果一致.单线态羟基卡宾反应物能量明显高于产物,产物更稳定;三线态反应物能量与产物能量相差较小,稳定程度相差不大.
图3(B)示出了单线态羟基卡宾分子及其衍生物质子转移过程的活化能垒示意图.可以看出,与羟基卡宾相比,强吸电子基团和强供电子基团使反应活化能增大,反应更难进行,强供电子基团使反应活化能增大幅度更大.—Cl,—Br等相对吸电性或供电性不强的取代基使反应活化能减小,反应更容易进行.因此,在单线态羟基卡宾质子转移反应中,强供电子基团和强吸电子基团均会使反应更难发生,吸电性或供电性相对较弱的基团会使反应更容易发生,—CH3使反应活化能降低最多,最容易反应.单线态羟基卡宾及其衍生物的产物能量比反应物低140.0~218.4 kJ/mol,所以产物更稳定.
图3(C)示出了三线态羟基卡宾分子及其衍生物分子质子转移过程活化能的示意图.可见,由于—CN具有极强的吸电性使反应活化能增大,其它取代基均使反应活化能下降;在单线态中具有吸电性的取代基使反应活化能增强,具有供电性的取代基使反应活化能减弱.因为单线态的电子是自旋相反的,而三线态2个电子自旋相同,三线态卡宾的过渡态的能量就更高一些,这就是单线态羟基卡宾的反应活化能比三线态低的原因.供电子基团促进电子在体系中流动,所以供电子基团更容易降低反应的能垒,促进反应进行.对于三线态,即便吸电子基团存在,也无法对活泼的电子产生影响,而—CN除外,因为—CN的吸电子能力极强.
对于三线态羟基卡宾分子及其衍生物这样的开壳层体系,其α电子密度分布和β电子密度分布不同,为了体现未成对电子在三维空间中的分布情况,将三线态羟基卡宾分子反应过程中反应物、过渡态和产物的自旋密度示于图S1(本文支持信息)中,将羟基卡宾分子衍生物的自旋密度图也示于图S1中.图中绿色代表正值,蓝色代表负值.由羟基卡宾分子异构化反应过程中的自旋密度变化图能够得到,反应物自旋密度正值主要集中在卡宾C原子区域,羟基区域中只存在很小一部分,其中部分衍生物的羟基O原子周围存在很少的一部分负值.反应过程中,C原子周围绿色区域逐渐向羟基上的O原子上转移,过渡态处H原子在C原子与O原子之间,H原子上的自旋密度为负值,产物处O原子周围电子绿色区域大于C原子周围,因此,三线态羟基卡宾及其衍生物质子化反应过程中,自旋密度正值由C原子逐渐向O原子周围转移.
2.2 PAEM和单电子作用势垒
2.2.1 羟基卡宾分子 图4(A)示出了1个电子在羟基卡宾分子平面HCOH(xy)上运动时,该电子所受到的作用势PAEM的三维立体图.在C,O和H原子核位置处,PAEM均有一个无限深的势阱,O和C原子核附近的势阱比H原子核附近宽大,一个重要的特征就是C与O原子之间的PAEM呈现一个马鞍形的曲面,该曲面鞍点处所对应的电子坐标使用1个五角星标出.该曲面与势能零的空隙间构成了C—O化学键的电子在2个原子间交流的通道,该通道的深度可以表征化学键的强度.图4(B)示出HCOH分子平面上PAEM的等值线图,靠近原子核的PAEM更低些,往外PAEM更高,红色线为所有单电子作用势与第一电离能负值相等的点构成的曲线,此线为该分子平面的MICC,成键的2个原子之间均有临界点,使用1个五角星标出.图4(C)为C—O化学键PAEM的鞍点示意图,该点处,沿着C—O化学键方向上有PAEM的极大值点,垂直化学键方向上有PAEM的极小值点,它们的交点就是鞍点,该点对应的PAEM的绝对值称为单电子作用势垒,用Dpb表示.图4(D)为沿着C—O化学键方向上PAEM随电子坐标的变化情况,在C与O原子间的PAEM有1个最大值点,此处PAEM的绝对值为该化学键PAEM的势垒(Dpb),此处所对应的电子坐标为C—O化学键的键心,用五角星标出.
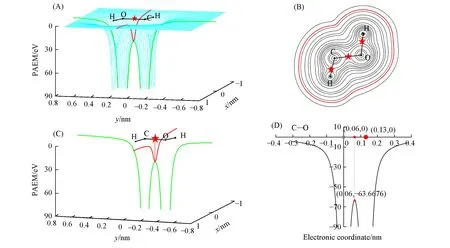
Fig.4 Three-dimensional graph(A)and the iso-PAEM graph of PAEM on the molecular plane of hydroxyl carbene(B),the diagram of saddle point between the C—O chemical bond(C)and PAEMcurve along the C—O chemical bond(D)
2.2.2 羟基卡宾分子质子转移反应过程 在羟基卡宾质子转移反应过程中,涉及O—H化学键的断裂,C—H化学键的形成,同时C—O单键转化为双键.在羟基卡宾分子质子转移反应中的IRC反应路径上,选取了9个点,1代表反应物,5代表过渡态,9代表产物,由反应物到过渡态选取了1~5个点,过渡态到产物选取5~9个点.按照前面所介绍的计算步骤,计算了每一个点所对应的羟基卡宾分子的PAEM,该羟基卡宾分子平面上的等PAEM图示于图5.从这个图中由反应物到过渡态可以看出,O—H化学键的逐渐断裂;由过渡态到产物为C—H化学键的逐渐形成.
Dpb是可以用来描述化学键强度的物理量[18],指在某个化学键区域内鞍点位置处的PAEM的绝对值,Dpb值的大小可以用来判断反应过程中化学键强弱的变化.使用上面表述的方法,在单线态羟基卡宾分子质子转移反应过程中,计算了C—O,O—H和C—H化学键的Dpb,列在表S2(本文支持信息)中.对于三线态羟基卡宾分子质子转移反应,也做类似的研究和探讨,计算的这3个化学键的Dpb也同样列在表S2中.图6绘出了化学键的Dpb与键长的相关图.

Fig.5 iso-PAEMdiagrams of the proton transfer reaction of singlet hydroxyl carbene along IRC route

Fig.6 Relationships between D pb and their respective bond lengths of the C—O(A),H—O(B),and C—H(C)chemical bonds in the process of proton transfer reactions of singlet and triplet hydroxyl carbenes
在单线态羟基卡宾反应过程中,C—O键键长与Dpb之间线性相关系数(R)为0.9908,在三线态羟基卡宾分子反应过程中R为0.9467,这两者之间有较好的线性关系[图6(A)],原子间距离越小,键长越短,Dpb值越大,键的强度越大.
反应过程中,从反应物到过渡态H—O键长逐渐增长,从过渡态到产物H…O间断键且距离逐渐增大.在单线态羟基卡宾分子反应过程中,从反应物到过渡态,H—O键长与Dpb线性相关系数R为0.9989,三线态的R为0.9996,所以单线态和三线态羟基卡宾反应过程中,H—O键长与Dpb之间也存在较好的线性关系[图6(B)].
反应过程中从过渡态到产物C—H键键长逐渐减小.在单线态羟基卡宾分子反应过程中,C—H键键长与Dpb线性相关系数R为0.9964,三线态的R为0.9883,所以单线态和三线态羟基卡宾反应过程中,C—H键长与Dpb之间也存在良好的线性关系[图6(C)].上述研究表明,Dpb值的大小可以用来判断反应过程中化学键的强弱变化.

Fig.7 Relationships between D pb and bond lengths of C—O chemical bond for singlet and triplet hydroxyl carbene and their derivatives
2.2.3 单线态和三线态的羟基卡宾分子及其衍生物的碳氧化学键 在单线态和三线态羟基卡宾及其衍生物的质子转移反应过程中,反应物C—O和O—H键长以及相应的Dpb值列于表S3和表S4(本文支持信息).图7为不同取代基对单线态和三线态羟基卡宾分子中C—O键键长以及Dpb值的线性关系图.由图7可知,不同取代基对羟基卡宾分子的C—O键长以及Dpb值有一定影响.但是C—O键键长与Dpb之间仍然存在线性相关,线性相关系数R为0.9771,距离越大,Dpb值越小,键的强度越小.在三线态羟基卡宾分子的结构中,不同分子C—O键键长与Dpb之间也存在线性相关,线性相关系数R为0.9429,由图可知,C—O键键长越长,Dpb值越小,键的强度越小.
2.3 羟基卡宾分子质子转移反应的分子形貌动态变化
图8为羟基卡宾分子质子化反应过程中反应物、过渡态和产物的分子形貌图,蓝色区域表示分子边界面前沿电子密度大,红色区域表示前沿电子密度小.从反应物的分子形貌图可以看出,C原子区域边界面的前沿电子密度为深蓝色,表示电子密度值大,与O原子相连的H原子区域边界面的前沿电子密度的颜色为深红色,说明电子密度最小.2个原子区域的界面电子密度的差促使了该反应发生,电子密度小的H原子向电子密度值大的C原子靠近,也就是电子密度小的区域向大的区域转移,即前沿电子密度指示羟基卡宾的质子转移反应,随着该反应进行,过渡态H原子向C原子靠近,C原子周围蓝色变浅,电子密度减小,O原子周围呈浅黄色,电子密度值增大,从过渡态到生成物,H原子不断靠近C原子,产物H原子与C原子相连,此时,O原子周围呈蓝色,电子密度最大.
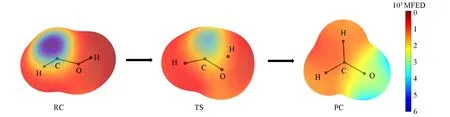
Fig.8 Molecular faces of reactants,transition states and products in H-transfer reaction the process of hydroxyl carbine
图9示出了反应的相对能量以及分子形貌的演变,选取单线态和三线态羟基卡宾反应路径上9个特征点,计算并绘出相应的分子形貌图.蓝色区域表示前沿电子密度值较大,红色区域表示前沿电子密度值较小.反应物与O原子相连的H原子周围红色最深,电子密度最小,C原子周围蓝色最深,电子密度最大;电子密度诱导H原子向C原子靠近,即反应作用是电子密度小的原子区域向电子密度大的原子区域靠近,这与SN2反应、马氏反应的规律一致.过渡态H原子向C原子靠近,C原子周围蓝色变浅,电子密度减小,O原子周围呈浅黄色,电子密度增大;产物H原子与C原子相连,此时,O原子周围呈蓝色,电子密度最大.
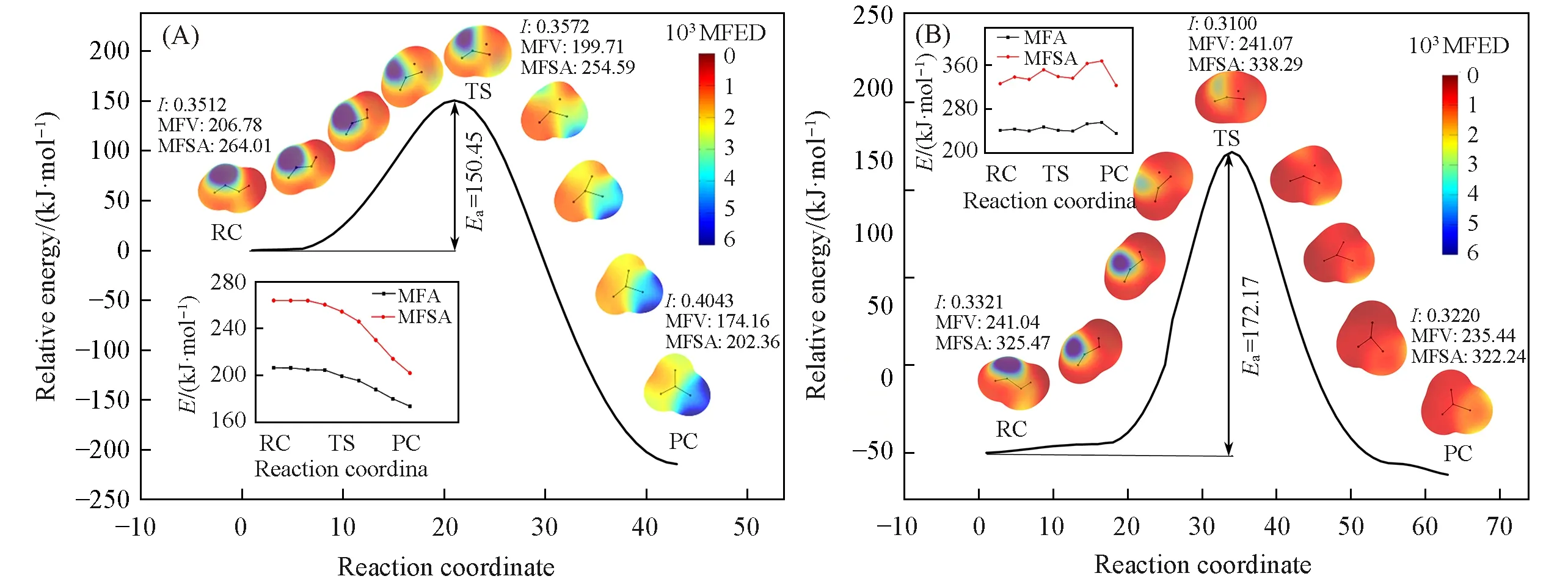
Fig.9 Molecular faces along the IRC reaction paths of proton transfer reactions of singlet(A)and triplet(B)hydroxyl carbenes
在沿着各自IRC反应路径上,单线态和三线态羟基卡宾分子质子转移反应的过程中,各自的分子形貌动态变化图分别示于图9(A)和(B)中.对于反应物,C原子区域电子密度最大,羟基上H原子区域电子密度最小,随着反应的进行,H原子逐渐远离O原子向C原子区域靠近,C原子周围电子密度逐渐减小,O原子周围电子密度逐渐增大,随着反应的进行,H原子与C原子相连后,O原子周围电子密度明显高于C原子,O原子周围电子密度最大.单线态反应过程中随着第一电离能逐渐增大,体积和表面积值逐渐减小,三线态反应过程中第一电离能先增大后减小,它们的体积和表面积先减小后增大,对比单线态和三线态羟基卡宾反应过程中的分子形貌图能够得到的是,在相同状态下,单线态分子形貌整体前沿电子密度高于三线态,单线态第一电离能大于三线态的,其体积和表面积小于三线态,面积小分子更紧密.
2.4 前沿电子密度与反应活化能的线性关联
羟基卡宾分子及其衍生物的前沿电子密度极大值出现在C原子区域,用符号表示.电子密度极小值出现在H原子区域,用表示;电子密度极大值与极小值的差,即C原子区域电子密度极大值与H原子区域电子密度极小值的差,用DED表示,这些数值列在表S5(本文支持确良信息)中,探讨了反应活化能与前沿电子密度的关系.C原子区域前沿电子密度值最大,除自身电负性极大的—F以外,其它取代基均使电子在该处的电子密度值减小;H原子处前沿电子密度值最小,—F,—Cl使电子密度极小值增大,其它取代基均使H原子区域的前沿电子密度极小值减小,这与取代基本身性质有关.
图10为前沿电子密度差值与反应活化能的线性相关图,由图10可知,DED与反应活化能之间存在一定的线性相关性,—H比较特殊,单独列出,对于—F,—Cl,—Br取代基对应分子的电子密度差值与反应活化能之间的线性相关系数为0.9992[图10(A)],—CN,—CHO,—OH取代基对应分子的电子密度差值与反应活化能之间的线性相关系数为0.9863[图10(B)],其它取代基线性相关系数为0.9823[图10(C)].可以看出,质子转移反应的活化能(Ea)与2个原子区域界面的电子密度差(DED)有线性关系,2个原子区域界面的前沿电子密度越接近,该反应的活化能越小,反应就越容易发生.
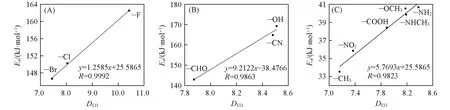
Fig.10 Relationships between D ED and activation energy(E a)of the proton transfer reaction of the singlet hydroxyl carbene and their derivatives
3 结 论
应用M06-2X/6-311++G(d,p)方法对单线态和三线态羟基卡宾及其衍生物(X=—H,—CN,—NO2,—CHO,—COOH,—COCH3,—F,—Cl,—Br,—CH3,—OH,—OCH3,—NHCH3,—NH2)进行优化,寻找反应过渡态并进行IRC验证,获得质子转移反应的反应活化能.计算了分子形貌理论的相关物理量,包括单电子作用势PAEM、Dpb、分子形貌、前沿电子密度等.研究和分析结果表明,具有吸电子和供电子的取代基对羟基卡宾分子的反应活化能的影响有一定规律性.在单线态中,强吸电子基团和强供电子基团使反应活化能增大,相对供电性或吸电性较弱的基团使反应活化能减小.三线态中,除吸电性极强的—CN取代基使反应活化能增大,其它取代基均使反应活化能降低.研究表明,分子形貌的前沿电子密度不仅是分子的基本特征,而且可指示或引导化学反应的位点和方向,甚至与反应活化能线性相关,这种规律性将进一步应用于更复杂反应的探索中.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20210211.
感谢美国华盛顿大学Davidson E.R.教授给予MELD从头计算程序方面的帮助.
