饲用植物甘草粗提物HPLC特征图谱的建立
2021-07-09刘梦婷钟晓红卿志星雷宝珠刘秀斌曾建国
■董 壮 1,2 刘梦婷 1,3 钟晓红 2 卿志星 1,3 雷宝珠 1 刘秀斌 1 曾建国
(1.湖南农业大学,中兽药湖南省重点实验室,湖南长沙 410128;2.湖南农业大学园艺学院,湖南长沙 410128;3.湖南农业大学动物医学院,湖南长沙 410128)
饲用植物是指农业农村部相关文件批准可以用于商品饲料生产的有一定应用功能的植物,具体产品表现形式有以干燥粉或粗提物为基础的饲料原料和以精提取物为基础的饲料添加剂[1]。甘草作为一种传统的中草药,近年来发现其具有很高的饲用价值。中药甘草为豆科植物甘草(Glycyrrhiza uralensis Fisch.)、胀果甘草(Glycyrrhiza inflate Bat.)或光果甘草(Glycyr⁃rhiza glabra L.)的干燥根和茎[2]含有多种化学活性成分,具有抗氧化、抗肿瘤、抗炎症、保护肝脏、改善心血管系统等一系列的重要功能[3]。同时甘草作为新型中草药饲料添加剂具有提升动物生长性能、增强机体免疫力、毒副作用小等优点,可以替代或者部分替代抗生素等化学合成添加剂,被广泛应用于山羊、绵羊、奶牛、家禽等的生产中,促进了畜禽生产性能的提高和机体的改善[4]。
随着市场对甘草及甘草粗提物需求的日益增加,野生资源的减少以及人工种植面积的增加,受地域、气候、海拔和土壤等因素影响[5],现有的甘草及其提取物的品质评价方法已经不能满足甘草的品质评价及资源综合利用的需要[6]。此外甘草作为新型的中草药饲料添加剂,其活性成分具有微量高效、成分复杂的特点,采用传统的价值评定系统难以确定其有效成分和作用效果。指纹图谱技术作为一种现代分析手段,常用以分析多维复杂问题,为饲料活性物质的研究提供新思路[7]。因此应用指纹图谱技术建立来自不同来源的34批甘草粗提物活性成分的特征图谱,能够从化学物质基础的角度考察甘草作为饲料添加剂的稳定性和可靠性,具有专属性、整体性、模糊性等特点[8],可以较为全面地反映甘草中所含主要活性成分的种类和数量[9]。甘草中所含化学成分主要以三萜皂苷类和黄酮类为主,还有香豆素、生物碱。根据已有文献报道,甘草中所含化学成分适宜使用高效液相色谱法(HPLC)测定[10],故研究按照《中国兽药典》通则中高效液相色谱法的要求建立分析方法,构建饲用植物甘草粗提物的特征图谱,为甘草的质量评价提供新的参考。
1 材料与方法
1.1 仪器
Waters 2695高效液相色谱仪(美国Waters公司);移液枪(德国Eppendorf公司);TG16-WS离心机(长沙湘智离心机仪器有限公司);KQ5200DE型超声波清洗器(昆山超声仪器有限公司);New Classic ME电子天平(METTLER TOLEDO);色谱柱Unitary C18(4.6 mm×250 mm,5μm)。
1.2 材料
无水乙醇(国药集团化学试剂有限公司生产,分析纯)、乙腈(色谱纯,Merck);超纯水(Milli-Q水系统)、磷酸(国药集团化学试剂有限公司,分析纯),甘草对照药材、对照品甘草苷(190726-009,≥98%)、甘草酸(190504-006,≥98%)均购自北京中兴嘉仁生物技术有限公司。
1.3 样品
从亳州、安国、廉桥三地的药材市场收集34批甘草样品,并对其编号。经鉴定,S10、S26为光果甘草,S13、S28为胀果甘草,其余均为甘草。详细采购信息见表1。
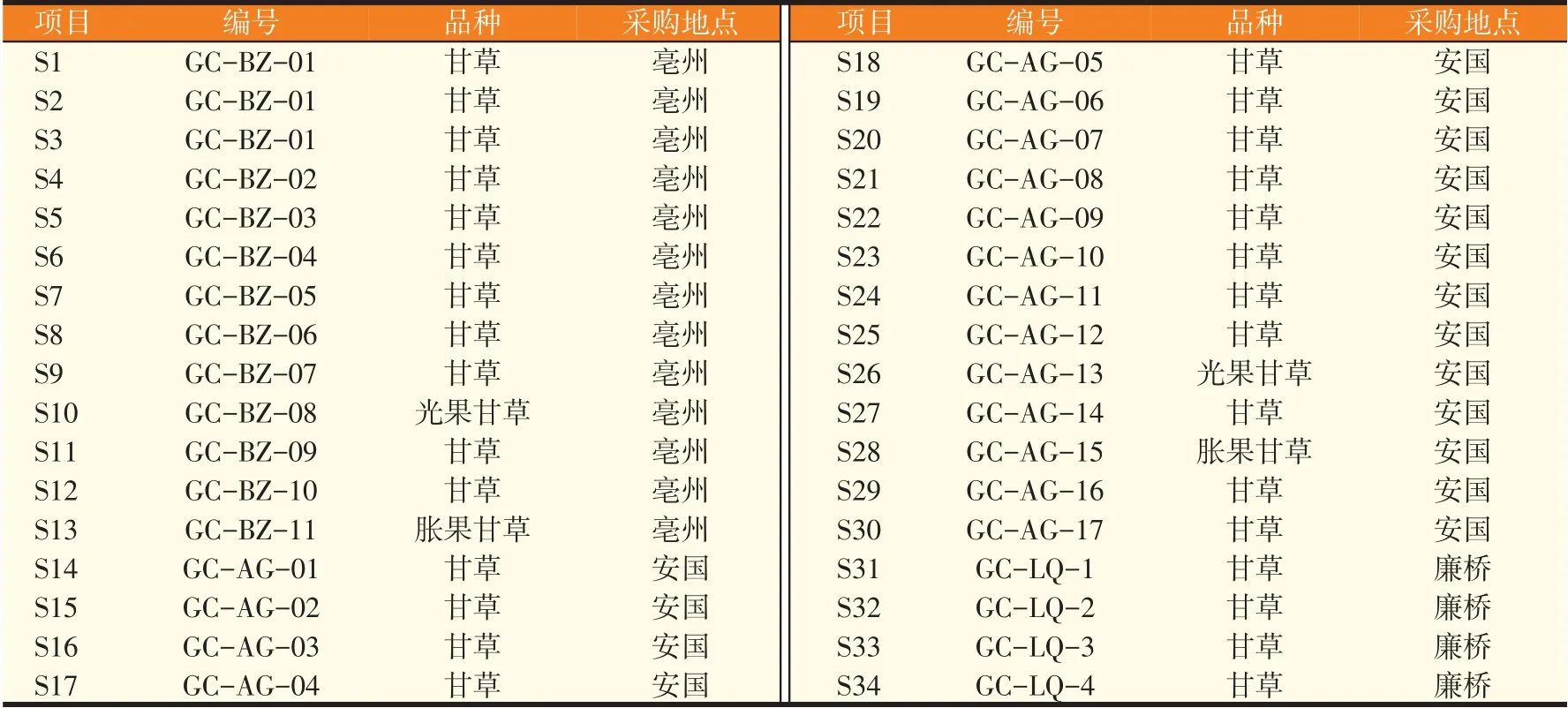
表1 34批甘草详细信息
1.4 色谱
1.4.1 色谱条件
色谱柱为Unitary C18(4.6 mm×250 mm,5μm);以乙腈(A)和0.05%磷酸水(B)为流动相,梯度洗脱,0~15 min,10%~25%A;15~35 min,25%~50%A;35~60 min,50%~90%A;45~60 min,70%~100%A;体积流量为1 mL/min;进样量为10μL;柱温为30℃;检测波长为276 nm;在以上的色谱条件下,各色谱峰分离效果最佳,基本达到基线分离。
1.4.2 样品的前处理
将上述收集到得甘草样品晒干粉碎,将甘草粉碎物过1号筛,取粉碎过筛后的甘草样品100 g,用70%乙醇回流提取2次,每次1 h,溶剂加入量依次为分别用500 mL和800 mL,合并提取液,滤液减压回收乙醇至稠膏状,60℃烘干,即得。
1.5 溶液的制备
1.5.1 供试品溶液的制备
称取样品粉末约0.20 g,置25 mL具塞锥形瓶中,精密加入乙醇-水(70∶30)25 mL,密塞,超声处理(功率400 W,频率37 kHz)30 min,取出,放冷至室温。取适量提取液于10 mL离心管中,12 000 r/min离心10 min;取上清液用0.22μm滤膜过滤,取续滤液于液相色谱小瓶中,备用。
1.5.2 参照物溶液的制备
分别精密称定甘草苷、甘草酸对照品各5.0 mg,置于25 mL棕色容量瓶中,加乙醇-水(70∶30)超声溶解定容至刻度,制成浓度为0.2 mg/mL的参照物储备液,于4℃冷藏备用。
1.6 方法学考察
1.6.1 精密度试验
取S1样品约0.20 g,精密称定,按“1.5.1”项下方法制备供试品溶液,按照“1.4.1”项下色谱条件连续进样6次,进样量为10μL,并记录色谱图。以3号峰为参照峰(S),8个共有峰的相对保留时间和相对峰面积的RSD分别小于0.2%和4%,说明该方法的精密度符合分析方法的要求。
1.6.2 稳定性试验
取S1样品约0.20 g,精密称定,按“1.5.1”项下方法制备供试品溶液,分别于0、1、2、3、4、5、6、12、24 h按“1.4.1”项下色谱条件进样,记录色谱图。以3号峰为参照峰,各特征峰以及S峰的相对保留时间和相对峰面积的RSD分别小于0.5%和3%,说明该方法稳定。
1.6.3 重复性试验
精密称定S1样品6份,每份约0.20 g,按“1.5.1”项下方法制备供试品溶液,按照“1.4.1”项下色谱条件进样,并记录色谱图。以3号峰为参照峰,各特征峰以及S峰的相对保留时间和相对峰面积的RSD分别小于0.1%和5%,说明该方法的重复性满足要求。
2 结果
2.1 特征图谱的建立
将34批甘草粗提物样品,按照“1.5.1”项下方法制备供试品溶液,按照“1.4.1”项下色谱条件测定,记录色谱图。采用仪器自带软件将所有色谱图叠加,根据叠加图谱中色谱峰分布,选择各批次共有色谱峰作为特征峰,建立统一处理方法处理各批次样品色谱图,确保每一批次特征峰均能被积分,再将色谱数据转化为*.AIA格式,将格式转化后的文件导入“中药色谱指纹图谱相似度评价系统(2012年版)”将“时间窗宽度”设定为0.2,进行多点校正。通过仪器自带软件和“中药色谱指纹图谱相似度评价系统(2012年版)”二次验证,共有特征峰在34批样品中均能识别,最后得到34批样品叠加图,并建立甘草粗提物特征图谱的对照图谱(R),详见图1和图2。通过两种方式,最终确定8个共有特征峰,按出峰时间依次标记为1~8号峰。以上指认特征峰均为甘草中有效成分,因此我们认为建立的特征图谱具有代表性,能对甘草粗提物进行质量控制[11]。

图1 仪器自带软件(Empower 3)对34批甘草粗提物的手动叠加图谱
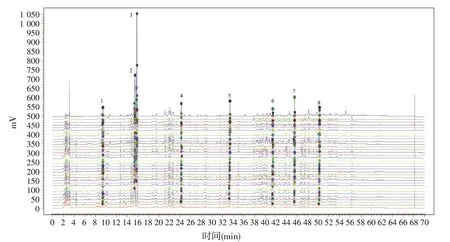
图2 34批不同来源的甘草粗提物的色谱叠加图以及对照特征图谱(R)
2.2 特征峰的指认
经文献查阅,参考药典,初步选定甘草苷、甘草酸为对照品,为考察特征图谱是否具有代表性以及能否表征待测所含成分的专属性,通过与对照品的HPLC谱图比对,明确保留时间为15.78、24.19 min的色谱峰分别为甘草苷(3号峰)和甘草酸(5号峰),采用质谱手段及对以往文献的查阅,明确保留时间15.38 min的色谱峰为芹糖甘草苷(2号峰)。鉴于甘草苷为甘草的主要有效成分,峰强度高,且保留时间适中,因此设定其为参照物(S峰);同时计算上述8个特征峰的相对保留时间(表2)和相对峰面积(表3)。结果显示,各批次样品特征峰的相对保留时间的RSD均在0.15%以下,说明特征峰出峰时间相对稳定,一定程度上体现了其质量的稳定性;而相对峰面积结果显示,各批次特征峰的峰面积差异较大,反映了不同来源批次甘草样品的差异性,由于甘草样品均采购自药材市场,因此无法准确从样品来源进行差异性分析。
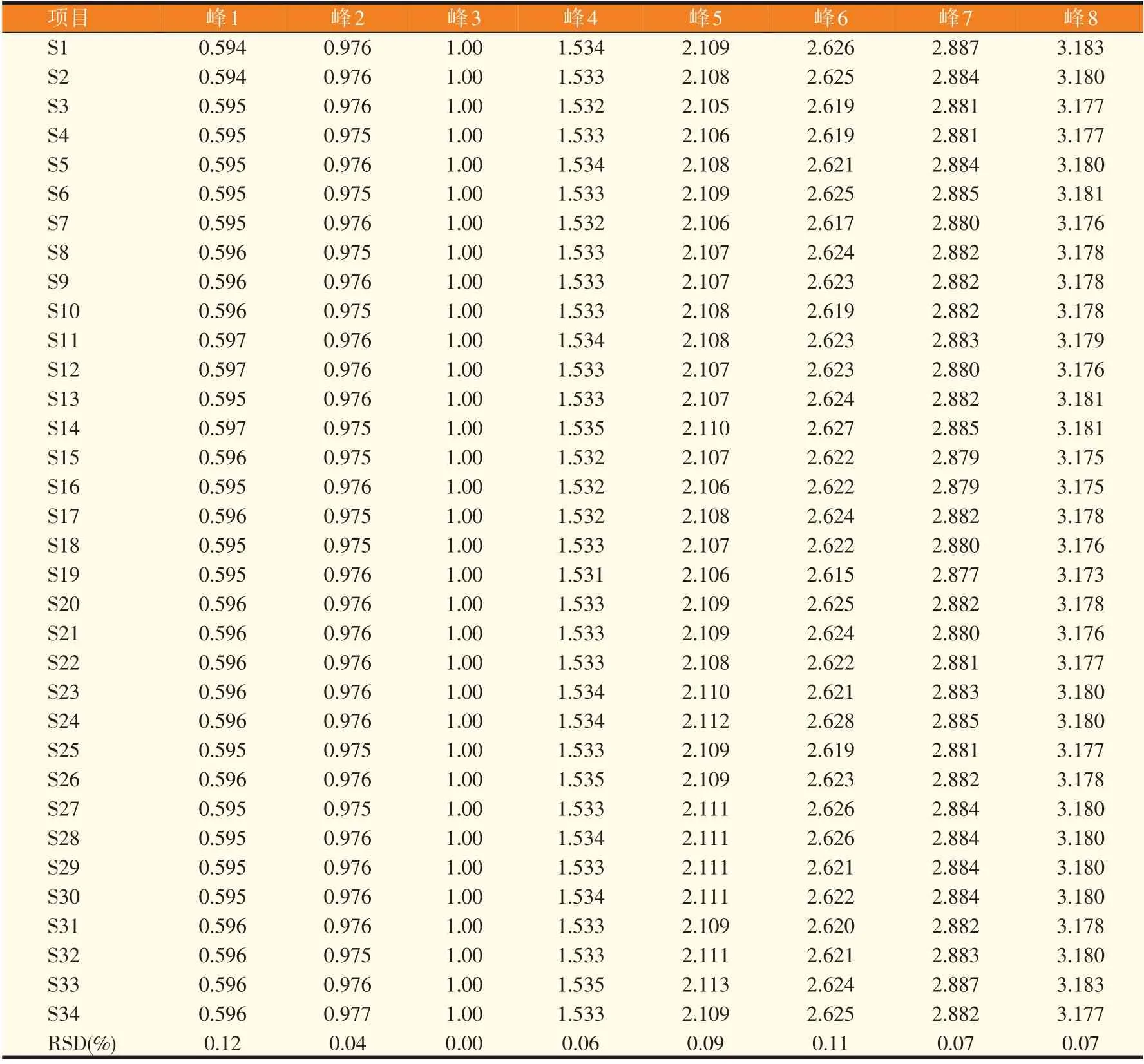
表2 34批甘草粗提物特征图谱中8个特征峰的相对保留时间
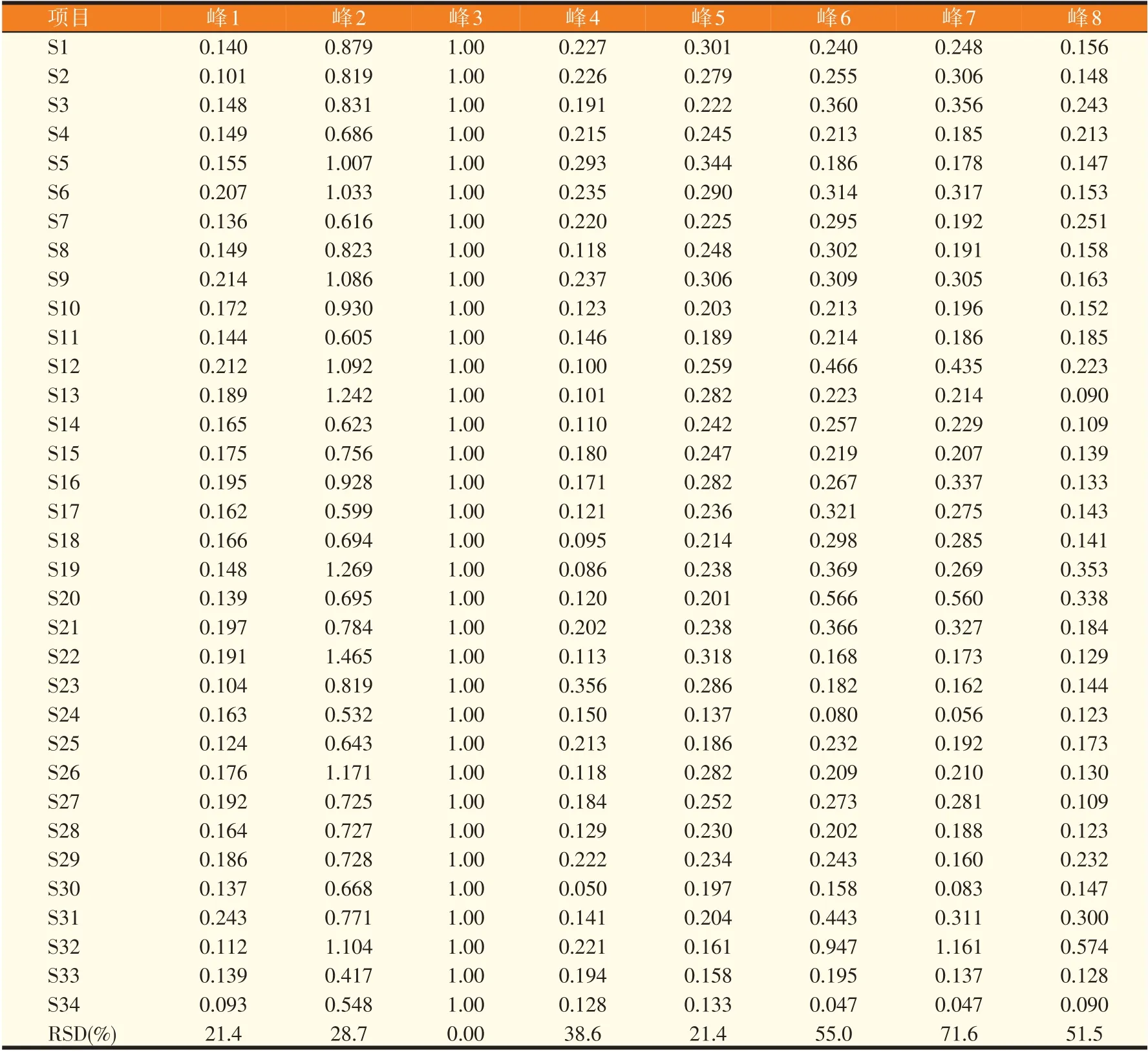
表3 34批次甘草粗提物特征图谱中8个特征峰的相对峰面积
2.3 聚类分析
将34批甘草粗提物样品的8个共有峰峰面积数据导入SPSS 26.0软件进行聚类分析,以平方欧式距离为测量区间,采用系统聚类的方法对数据进行分析[12]。结果显示:34批样品(除S32外)被分成4大类(如图3)。S6、S9、S13、S22、S26为一类,S1~S2、S5、S23为一类,S3~S4、S7~S8、S10~S11、S14~S18、S21、S24~S25、S27~S28为一类,S12、S19~S20、S31为一类。由此分类可知,甘草品质不具有明显的地域性,尽管来自多地,但是内在品质较为均一。
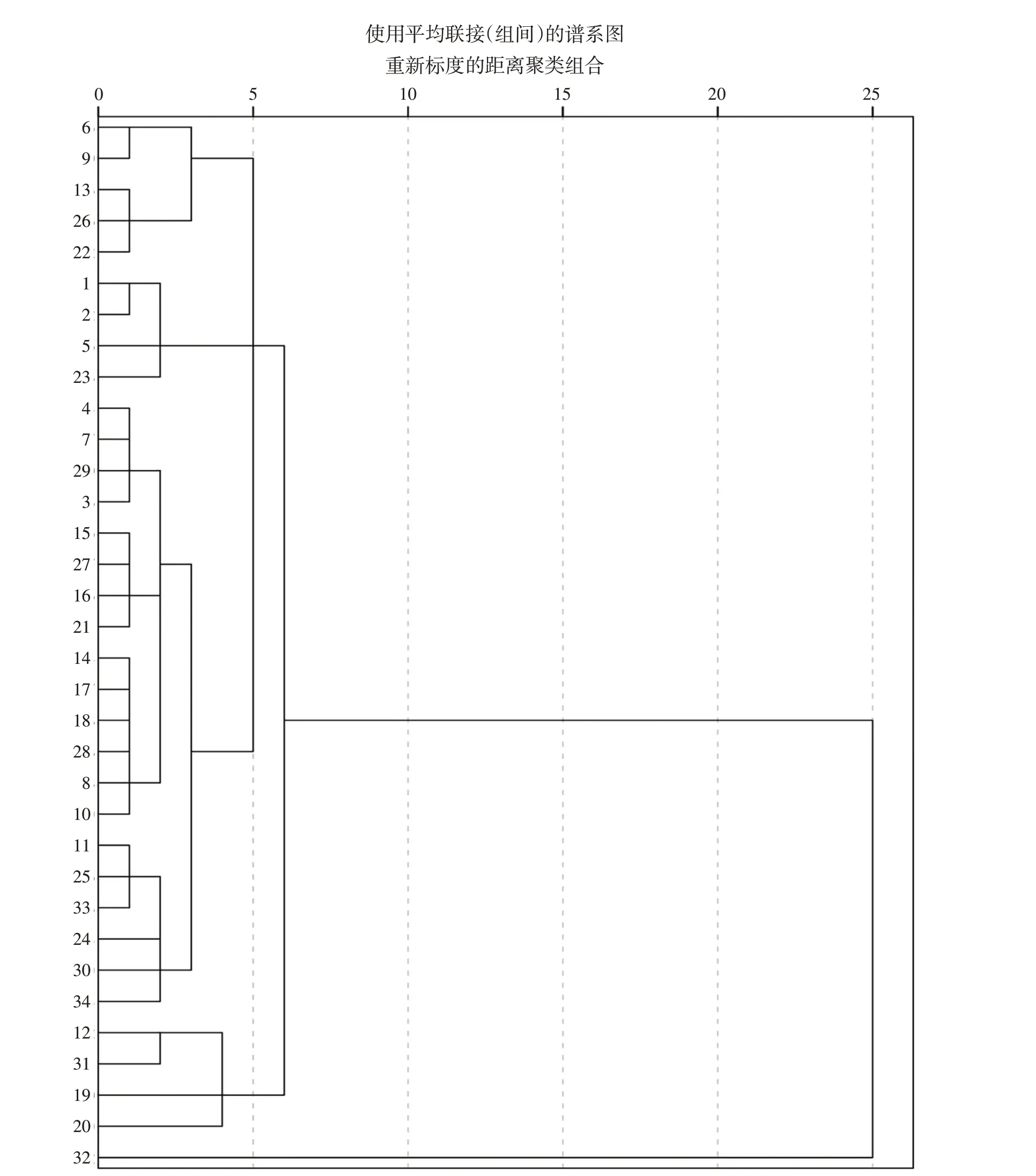
图3 聚类分析
3 讨论
3.1 前处理优化
为确保甘草粗提物主要成分最大限度地转移到溶液中,便于样品测定。由于甘草粗提物是经70%乙醇回流提取、浓缩、干燥而得,其主要成分均易溶于70%乙醇中,因此选用70%的乙醇作为溶剂,并对溶剂的用量和提取方式进行考察,三种不同提取方式分别为0.1、0.2、0.5 g甘草粗提物用25 mL 70%的乙醇提取。最终确定0.2 g甘草粗提物经25 mL 70%乙醇超声30 min,可全部溶解。
3.2 分离方法的优化
3.2.1 色谱柱的选择
取供试品溶液,分别考察了Unitary C18(4.6 mm×250 mm,5μm)、Sunfire(4.6 mm×250 mm,5μm)、Di⁃amonsil(4.6 mm×250 mm,5μm)三根色谱柱对甘草中主要色谱峰分离的影响,评估了分离度、峰形、出峰时间等因素,结果表明不同色谱柱对分离效果影响不大,故该试验选择Unitary。
3.2.2 检测波长的考察
本试验采用二极管阵列检测器(PDA)对样品进行全波长扫描,检测结果多数色谱峰的最大吸收波长为276 nm,且在最大吸收波长下样品测定结果中色谱峰数量及响应程度均优于其他波长,因此选择276 nm作为数据采集波长。
3.2.3 流速的考察
流动相的流速越大,被分离物质出峰时间越快,响应高;较小的流速会造成分析时间过长,色谱峰展宽,造成拖尾、响应变低等后果。考察不同流速对甘草粗提物中主要有效成分分离的影响,结果表明流速为1.2 mL/min时有部分成分未能完全分离,0.8 mL/min时分离效果与1.0 mL/min相近,但分离时间过长,故该流速为1.0 mL/min。
3.2.4 柱温和的考察
考察柱温25、30、35℃对甘草粗提物中主要有效成分分离的影响,结果表明不同柱温对分离效果影响不大,故综合考虑选择柱温30℃。
3.2.5 进样量的考察
考察进样量为3、5、10μL对甘草有效成分分离的情况,结果表明进样量为3μL和5μL时甘草粗提物有效成分色谱峰响应偏低,故选择10μL作为本试验的进样量。
4 结论
本次试验通过对前处理方法和仪器条件进行优化,建立了良好的HPLC分析方法,使得甘草各化学成分在得到良好的分离,结合指纹图谱技术对34批来自不同产地的甘草粗提物进行了分析,得到了其容易识别、分离度较好的特征图谱,各批次样品图谱中共包括8个特征峰,以3号峰为参照峰,同时计算共有峰的相对峰面积,并对结果进行了聚类分析。通过与对照品色谱峰比对,并结合质谱手段以及文献查阅,确定了色谱峰2、色谱峰3和色谱峰5分别为芹糖甘草苷、甘草苷和甘草酸。本次研究构建的34批不同产地的甘草粗提物的特征图谱具有良好的精密性、重复性和稳定性,可用于构建基于产地差异的饲用植物品质评价方法,是一种更高效的质量控制的方法。此外,建立的HPLC分析方法结合指纹图谱技术作为一种现代检测手段,基本可实现对甘草中活性物质的整体描述与评价,另外可在判别甘草的真伪优劣以及综合利用等方面提供一定的参考价值[13]。
