电解液化学保护锂金属电池负极的研究进展
2021-07-08林元华陈杨阳冯炫杰纪洪江王明珊陈俊臣
杨 珺,林元华,廖 丽,陈杨阳,冯炫杰,李 佩,纪洪江,王明珊,陈俊臣,李 星
(西南石油大学 新能源与材料学院,成都 610500)
从1800年伏达(Volta)发明了伏达电堆以来,已经有多种电池体系被人们商业化,包括铅酸电池、镍镉电池、镍-氢电池以及锂离子电池[1]。锂离子电池无疑是其中最具代表性的。如今,锂离子电池广泛应用于各类便携设备中,并在下一代新能源车型中被大力推广,化学储能电源在当今社会中不断凸显出明显的市场地位。但是,即使锂离子电池经过了数十年的发展,其能量密度(≈250 Wh/kg,单体电池)依然难以满足人们的需要[2]。高活性锂金属因具有低的氧化还原电势(-3.04 V对标准氢电极)及高的理论比容量(3860 mAh/g),被认为是综合性能最优异的负极材料。图1为不同电池体系的能量密度对比,使用锂金属作为负极的Li‖LMO(过渡金属氧化物正极)电池能够直接达到约440 Wh/kg的能量密度[3]。在Li-S,Li-O2电池中,更是能够直接达到2600,3505 Wh/kg的能量密度[4]。近年来对锂-硫电池、锂-空气电池研究的不断深入,加快了锂金属电池的应用速度[5]。然而,锂金属作为负极时存在低库仑效率及严重的锂枝晶生长问题并未从根本解决,前者会造成电池容量及循环性能的迅速衰减,后者会给电池带来严重的安全隐患。
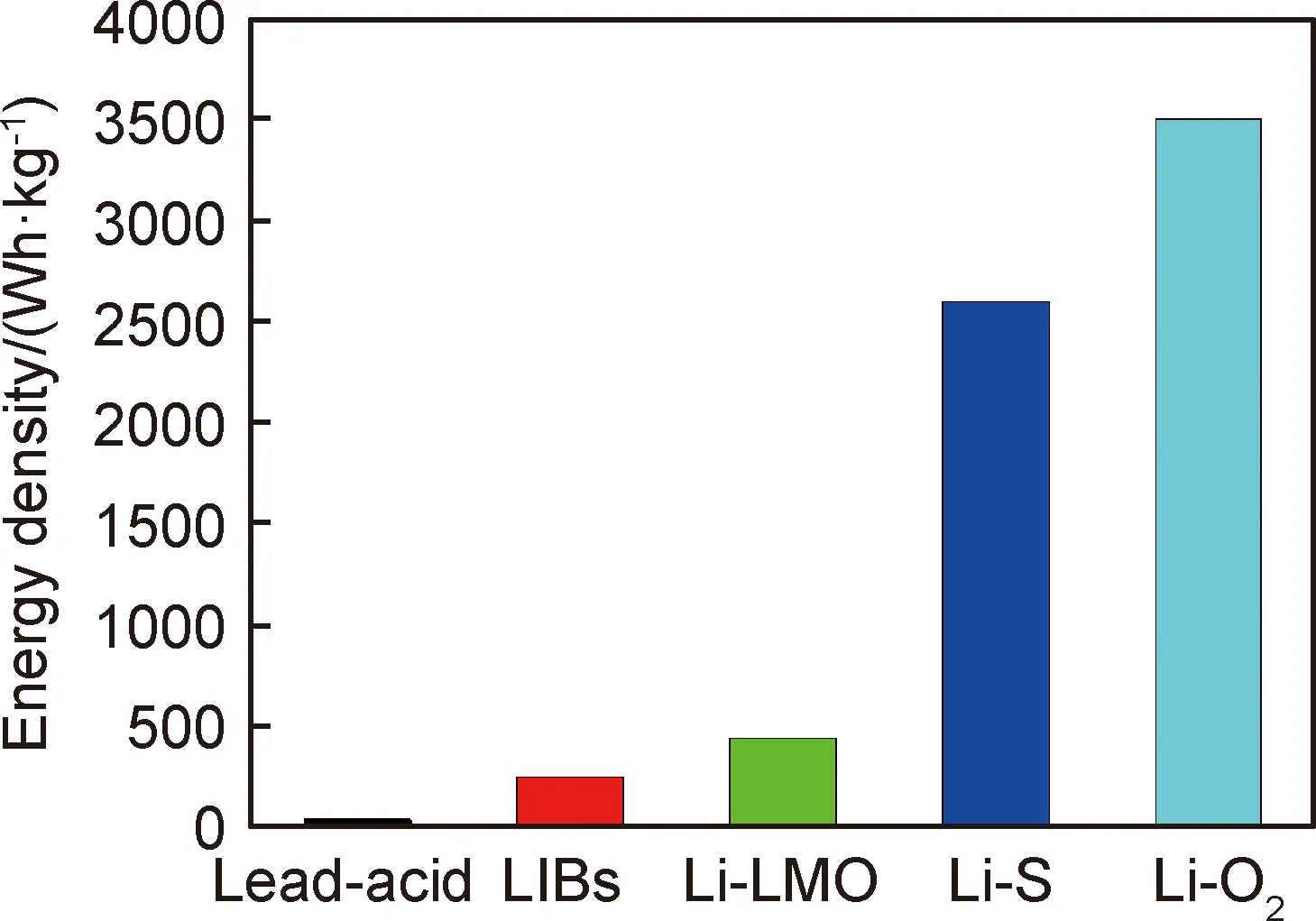
图1 各种电池体系能量密度[3]Fig.1 Energy density of various battery systems[3]
对锂金属负极的研究可以追溯到20世纪60年代,当时美国军方在考虑登月使用的电源时,想到了锂金属作为负极的非水电解质电池。但是,由于安全性问题一直不能解决,锂金属负极的研究一度被搁置。近年来人们对高能量密度电池提出了更高要求。此外,随着表界面科学的快速发展,科学家们对操纵表界面技术有了更多的自信,综合性能优异的锂金属负极又重新被人们关注并成为近10年来研究的热点。
1 锂枝晶生长机制
和大多数活性金属材料一样,锂金属在电沉积过程中易产生锂枝晶。锂枝晶的形成会刺穿隔膜使电池内部短路,是锂金属电池最大的安全隐患。锂金属极低的氧化还原电势(-3.04 V对标准氢电极)使其具有强还原性,易与电解液发生反应。与电解液反应产生的不溶性副产物将积累在锂金属表面形成固态电解质界面膜(SEI),SEI膜是一种只允许锂离子通过而不允许溶剂等大分子通过的电子绝缘界面膜。锂枝晶的生长将导致更大表面积的锂金属暴露在电解液中,这些新形成的热力学不稳定的锂金属表面将快速和电解液中的组分发生反应,导致电解液持续消耗,最终使电池提前失效。同时,在锂金属表面反复沉积/剥离过程中将使锂金属的体积发生较大改变,如果SEI膜的强度或者韧性不足,将导致SEI膜破裂,使电解液与活性锂金属表面重新接触、反应,SEI膜持续增厚。此外,锂金属在反复的沉积/剥离过程中还可能形成大量“死”锂层(被SEI膜包覆,不能继续参加电化学反应的金属锂孤岛),“死”锂层的形成不仅同样暴露出更大的锂金属表面,同时还会消耗大量活性锂金属。锂枝晶生成会给锂金属电池带来严重的安全隐患,不断增厚的SEI膜与“死”锂会使锂金属电池呈现出低的库仑效率,严重影响电池容量及循环稳定性。
锂枝晶的形成与锂金属负极的结构形貌、沉积/剥离电流密度/电荷量、电解液的化学组成以及锂盐的浓度、工作温度等密切相关。经过数十年的研究,人们总结出了多种锂枝晶的生长模型,如:空间电荷模型、表面形核与扩散模型、Sand’s time模型等。但由于锂枝晶的生长条件极其复杂,至今未能成功建立一个普遍适用的模型[4]。
锂金属电池电解质中锂离子的浓度以及锂离子的扩散效率是导致锂枝晶形成的关键因素。在锂沉积过程中,沉积界面附近电解质中的锂离子不断被沉积到锂金属表面,然后由从正极扩散过来的锂离子继续补充。电沉积过程中,两电极间将存在一个浓度梯度,当电流密度达到一个临界值J*时,在Sand’s time时间“τ”后将不能再维持该电流密度,此时锂金属表面区域电解液中阳离子被消耗完,打破了表面层的电中性,从而出现空间电荷,促进树枝晶锂的形核生长。因此适当提高电解液中锂离子的浓度在一定程度上能够有利于抑制锂枝晶的形核生长[5-7]。
上述理论模型简要地指明了锂枝晶的生长过程,但是锂金属的高反应活性使得其在沉积过程中极大地受到表面化学的影响。由于锂金属的高反应活性,其与电解液接触时立即发生反应,产生不溶性产物形成SEI膜。SEI膜的质量也会严重影响锂枝晶的生长。在锂金属沉积/剥离的过程中,由于SEI膜组成不均匀,锂金属表面Li+扩散能垒不一致,从而促使锂金属不均匀形核[8]。同时在锂金属的沉积/剥离过程中的体积改变导致SEI膜破裂,破裂处的锂金属表面具有更低的Li+扩散能垒,这也会使锂金属在SEI膜破裂处优先沉积,产生锂枝晶[9-10]。优质的SEI膜应该具有较高的表面能以提供足够的力学性能适应在锂金属沉积/剥离过程中的形状体积改变,同时需要较低的锂离子扩散能垒[11]。电解液的化学组成和锂盐浓度对SEI膜的质量密切相关。
锂枝晶的生长机制仍有待进一步深入研究,由于SEI膜复杂的成分和锂枝晶对电子束的敏感性,利用冷冻电镜技术是研究锂枝晶生长的有效途径[12-16]。通过表界面技术、电解液化学等途径,尽可能消除或者抑制锂枝晶在锂金属负极的生长是提高锂金属电池安全性、推动其实用进程的关键。
2 锂金属负极保护
为了保护锂金属负极,人们尝试了多种途径,如电解液化学、锂金属表面修饰、固态电解质、负极微结构设计、锂负极合金化等。其中,电解液化学被认为是最简便,成本最低、最容易实现产业化应用的一种途径。
电解液作为电池的主要组成部分之一,在锂金属电池中起到传输锂离子以及生成稳定SEI膜的作用,电解液对锂金属电池的阻抗、循环稳定性、枝晶生成、库仑效率等具有决定性影响。
2.1 溶剂
酯类因其具有较高的抗氧化性能以及较好的低温锂离子导电性能而成为一种备受关注的电解液溶剂。现今阶段,酯类电解液被广泛应用于锂离子电池中,碳酸亚乙酯(EC)在锂离子电池中的应用极大地抑制了溶剂分子共嵌入石墨负极。在锂金属电池中,EC在少量路易斯酸五氟化磷(PF5)的催化下能够聚合产生具有优异力学性能的稳定SEI膜[17]。具有较高介电常数的EC更有利于锂盐解离,提高锂离子迁移数。而具有较低黏度的碳酸二乙酯(DEC)能够通过降低电解液黏度来提高锂离子迁移率,因此通常将EC与DEC用作混合溶剂。EC/DEC/FEC(氟代碳酸乙烯酯)作混合溶剂的高浓度(2 mol·L-1)LiPF6电解液在无负极Cu‖NMC333电池中以97.8%的库仑效率(CE)循环50次后依然剩余超过40%的容量[18]。在0.6 mol·L-1LiDFOB(二氟草酸硼酸锂)+0.6 mol·L-1LiBF4(四氟硼酸锂)-FEC/DEC(1∶2体积比)电解液中,锂金属沉积形貌为粗大均匀的无枝晶形貌(1200 kPa压力下),同时具有超过4.5 V的高压性能[19]。酯类高浓度电解液(LiFSI(双氟磺酰亚胺锂盐)∶DMC(碳酸二甲酯)=1∶1.1)具有超过5.2 V的超高电压窗口[20],同时在高浓度锂盐的作用下能够在锂金属表面生成稳定的SEI膜,其在Li‖Cu电池中的库仑效率高达99.2%[21]。然而,酯类溶剂的抗还原性能相对醚类更差,在Li‖NMC811电池中对比LiFSI基DMC与DME(乙二醇二甲醚)高浓度电解液的循环性能时,醚类(DME)高浓度电解液明显具有更加优异的循环性能[22]。由于酯类本身较低的抗还原性能,在应用于锂金属电池中时更需要采用成膜添加剂或者高浓度等策略辅助成膜。
TGF-β(transforming growth factor beta,转化生长因子β)是一种多功能的多肽类细胞因子,哺乳动物组织中存在3种TGF-β亚型:TGF-β1、TGF-β2和TGF-β3,其中TGF-β1最为重要。几乎体内的所有细胞都能产生TGF-β并存在其受体,参与细胞的分化、增殖、转移、形态的改变以及凋亡等,在机体的免疫调节、细胞生长和分化、肿瘤免疫等发面均起到了重要的作用。人类TGF-β1基因定位于染色体19q3,含有7个外显子,其5ˊ端序列含有5个调控区:1个类增强子活性区,2个负调控区和2个启动子区。
根据分子轨道(MO)理论,拥有相对更低的最高占据分子轨道能(HOMO)的分子具有更强的抗氧化能力,拥有相对更高的最低未占用轨道能(LUMO)的分子具有更强的抗还原能力。图2是根据分子轨道理论计算出各类非质子溶剂的轨道能量。其中醚类普遍具有更强的抗还原能力,意味着醚类是和锂金属更兼容的溶剂[23]。

图2 部分电解液溶剂的HOMO与LUMO能Fig.2 HOMO and LUMO energies of some electrolyte solvents
DME具有线性醚类中最低的还原电位,Park等[24]研究多种有机溶剂对锂金属对称电池的稳定性,分析了电解液溶剂、黏度、阴离子大小对锂金属稳定性的影响。研究发现,锂金属电解液应该具有以下特征:溶剂应具有较低的还原电位,盐阴离子应具有较大的体积,得到的电解质应具有较低的黏度。在使用DME作锂金属电解液溶剂与1 mol·L-1双三氟甲基磺酰亚胺锂(LiTFSI)锂盐配制的电解液中Li‖Li对称电池能够在3 mA·cm-2电流密度以及12 mAh·cm-2的大沉积容量下稳定循环超过100次(800 h)[24-25]。
1,4-二恶烷(DX)具有环醚中最低的还原电位[24],意味着DX溶剂与锂金属具有更好的兼容性,在1 mol·L-1双氟磺酰亚胺锂(LiFSI)-DX/DME电解液中Li‖Li电池稳定循环时间是使用纯DME溶剂的4倍以上。同时,使用DX-DME混合溶剂的电解液的氧化电位(≈4.78 V)明显比纯DME溶剂的电解液的氧化电位(<4 V)更高。采用1 mol·L-1LiFSI-DX/DME能够在较低的电解液浓度下达到高电压的目的。这主要是抗氧化能力较强的DX出现在溶液中,迫使相对更多的锂盐阴离子(相对更少的溶剂分子)被氧化,产生富含LiF的致密均匀且电导率更高的SEI钝化层[26]。
1,3-二恶烷(DOL)由于其α位的H原子易于被氧化,在将其所有位点处的H原子由甲基基团取代之后形成2,2,4,4,5,5-六甲基-1,3-二恶烷(HMD),其具有更稳定的抵抗过氧化物或者单个氧原子袭击的能力,结合硼酸辅助形成SEI膜时,其在Li-O2电池中具有超过普通溶剂(DME,DOL)4倍的性能表现[27]。
实际上,醚类电解液虽然具有对锂金属更好的兼容性,但是其较低的抗氧化性能通常限制了其在高压锂金属电池中的应用。因此在高压(>4 V)范围内,常规醚类电解液较少应用。
2.2 氟代溶剂
因为F原子的强吸电子效应,通常使得氟代溶剂具有较高的抗氧化性能,是一种用于高压电解液的备选材料。同时,氟代溶剂能够为SEI膜提供F源,利于产生高氟化锂(LiF)含量的SEI膜。三氟乙基甲基碳酸酯(F-EMC),1,1,2,2-四氟乙基-2,2,3,3-四氟丙基醚(F-EPE)等氟化溶剂配合六氟磷酸锂(LiPF6)配制出5 V以上的高压电解液[28]。FEC是一种对锂金属较温和的溶剂,当使用FEC作7 mol·L-1LiFSI电解液溶剂时能够使锂金属电池具有超过5 V的高压性能,并能帮助在锂金属表面生成高LiF含量的SEI膜。Li‖Cu电池超过99%的高库仑效率(CE)证明其能够与锂金属保持高度稳定[29]。氟代溶剂除了具有高压特性外,同样能够提高锂金属负极的库仑效率。LiPF6溶解在FEC,FEMC,HFE(1,1,2,2-四氟乙基-2,2,2-三氟乙基醚)(质量比2∶6∶2)全氟代溶剂形成的电解液,在Li‖Cu电池测试时,锂金属库仑效率高达99%。当使用全氟代溶剂电解液时,锂金属表面SEI膜中具有超高LiF含量(≈90%)。LiF在SEI膜中不仅是良好的电子绝缘体,同时其较低的Li+扩散能垒有助于锂离子扩散通过SEI膜,此外,其较大的表面能还赋予SEI膜优异的力学性能。在全氟代溶剂电解液中易于形成粗大无枝晶的锂沉积形貌[30-32]。高浓度LiFSI锂盐电解质在锂金属电池中具有优异的性能,这主要是其FSI-阴离子分解形成了稳定的SEI膜[33-35]。氟化氨磺酰(FSO2NC2H6:FSA)是一种灵感源于FSI-的溶剂,其具有FSI-类似的结构。在使用FSA作LiFSI的溶剂时能够在稀盐电解液中达到优异的性能,Li‖Cu电池CE可达99%,并能够产生致密、平整、富含LiF的SEI膜[36]。氟化溶剂的应用打破了常规溶剂电压限制的同时,为SEI膜提供了更多F源,利于产生富含LiF、更加稳定的SEI膜。
2.3 锂盐
锂盐在锂金属电解液中除了提供锂离子之外,其阴离子还大量参与成膜。锂盐对锂金属负极SEI膜的质量起到至关重要的作用。图3是部分电解液常用锂盐HOMO与LUMO能,双草酸硼酸锂(LiBOB)因具有明显更高的HOMO值与相对更低的LUMO值,使其通常最先在电极上发生反应形成稳定的SEI膜,但是LiBOB溶解度低,限制了其更广泛的应用。单一锂盐,特别是稀盐电解液,其性能经常难以满足人们的需要。通过把两种或者几种锂盐联合使用在一定程度上能够提高锂金属电池的库仑效率。例如,在综合分析LiTFSI,LiFSI,LiBOB,LiDFOB四种锂盐两两组合溶解在EC/EMC (质量比4∶6)形成的双盐电解液时,通过理论计算与实验数据发现其电化学性能呈现如下趋势:LiTFSI-LiBOB>LiTFSI-LiDFOB>LiFSI-LiDFOB>LiFSI-LiBOB。LiTFSI-LiBOB复合锂盐能够帮助在锂金属表面产生稳定致密的SEI膜[37]。在醚类(DME)电解液中使用高浓度LiTFSI-LiDFOB复合锂盐也能够明显提高锂金属电池性能[38]。LiTFSI-LiFSI/DME电解液具有比单盐电解液更稳定的性能[39]。冷冻透射电镜(Cryo-TEM)表明,在LiPF6碳酸酯类电解液中锂枝晶呈细小且杂乱的丝带状,该沉积形貌具有大的表面积使锂金属与电解液发生更多副反应,且锂金属负极的库仑效率不高。高浓度的LiFSI(4.6 mol·L-1)电解液中的锂枝晶则具有纳米片状和丝带状的混合结构。在超高浓度的LiTFSI-LiFSI(7 mol·L-1)双盐电解液中的锂沉积形貌明显呈现出均匀的纳米片状结构[40]。

图3 部分常用锂盐HOMO与LUMO能Fig.3 HOMO and LUMO energies of some lithium salts
不同锂盐的混合使用往往能够发挥出令人惊奇的效果。例如,LiDFOB是一种优质成膜锂盐,但是其在4.3 V以上将会分解产生大量气体,这与其本身较高的HOMO值有关(如图3所示)。在使用0.6 mol·L-1LiDFOB +0.6 mol·L-1LiBF4混合锂盐时(FEC/DEC),电池在高电压循环时产气量大幅度下降,在Cu‖NMC532电池中(外加堆叠压力1200 kPa),在4.5 V下稳定循环100次后容量保持率仍能达到80%,且锂金属沉积形貌为无枝晶的结节状[19]。这些实例进一步说明了双盐电解液在抑制锂枝晶、提升锂金属负极库仑效率方面具有明显的优势。
2.4 稠盐电解液
锂盐浓度严重影响电解液的离子电导率、黏度以及所对应的电池稳定性。目前商业化的电解液中通常采用1 mol·L-1LiPF6用作电解液的溶质,但是对于锂金属电池来说,发展高浓度电解液可能是潜在发展方向。在7 mol·L-1LiTFSI-DOL/DME(1∶1)电解液中具有明显高的锂离子迁移数(0.73),将其应用在Li-S电池中能够具有优异的稳定性。由于超高浓度电解质中的溶剂分子与电解质盐的阴阳离子络合作用已达到饱和,使其不能再溶解多硫化物,于是多硫化物的穿梭效应被明显抑制。同时高浓度电解质具有更高的锂离子迁移数,在锂金属沉积过程中提供更加均匀的锂离子流,因此高浓度电解液中具有比低浓度的同类型电解液明显均匀的锂沉积形貌[33]。
人们在对醚类电解液探索中发现当溶剂分子与锂离子形成离子鞘后能够有效降低分子的HOMO值,提高电解液的抗氧化性能[43-44]。对于醚类电解液,提高其浓度能够有效增加其抗氧化能力,使其能够结合高电压正极材料使用。同时高浓度电解液中具有更高的锂离子浓度和锂离子迁移数使其具有明显高的倍率性能[45],4 mol·L-1LiFSI-DME电解液的Li‖Li对称电池能够在10 mA·cm-2的电流密度下稳定循环超过6000次,Li‖Cu电池保持99.1%的超高库仑效率稳定循环1000次。该电解液的线性扫描伏安法(LSV)测试表明其氧化电位超过4.5 V,明显高于同类型稀盐电解液(<4 V)[34]。在磷酸铁锂无负极(LiFePO4‖Cu)电池中,该电解液循环100次平均库仑效率为99.8%,循环50周次后容量保持率为60%,性能远高于六氟磷酸锂基碳酸酯类电解液[35]。进一步应用到三元正极锂金属电池中时,该电解液在4.5 V电压下亦可稳定循环。对电池正极表面阴极电解质界面膜(CEI)进行光电子能谱(XPS)分析表明,高浓度LiFSI电解液能够在正极表面形成富含F(28%(原子分数,下同)~35%)的薄(5 nm)而致密的CEI。此CEI膜能够有效保护正极材料不被电解液腐蚀。同时在对比DME与DMC两种溶剂下的高浓度LiFSI电解液时,DME类电解液长循环性能明显优于DMC[22]。当2 mol·L-1LiTFSI-2 mol·L-1LiDFOB溶解在DME形成高浓度的电解液被应用到Li‖NCM333(负载量10.8 mg·cm-2)电池中时,在0.3 C充电、1 C放电倍率下此高浓度双盐电解液亦能提供超过500次的稳定循环(4.3 V循环500次后容量保持率79%),但此电解液的Li‖Cu电池对应的锂金属库仑效率较低(94%)[38, 46],明显低于4 mol·L-1LiFSI-DME电解液(99%)[34]。高浓度电解液中更高的锂离子迁移数能够提供更大的临界电流密度,提高电解液浓度能够在一定程度上抑制锂枝晶生长。
对于LiTFSI与LiFSI两种锂盐来说,此类稀盐电解液在4 V以上电压时将对正极铝集流体造成严重腐蚀[47-48],但是当浓度足够高时(>3 mol·L-1)对Al集流体的腐蚀能够得到抑制。一般认为是高浓度电解液中溶剂分子与锂盐阴阳离子饱和配对,使得电解液对铝集流体的腐蚀产物不能再继续溶解,迫使其在铝集流体表面累积并最终停止其后续腐蚀反应[20,34,38,49]。
除醚类溶剂之外,在其他一些溶剂中采用高浓度的策略也可能会收获优异的效果。高浓度电解液中含有大量锂盐阴离子,当阴离子的量足够多时,电解液与锂金属反应主要为阴离子被还原,这将更有利于产生稳定致密的SEI膜,因此高浓度电解液能够通过生成更加稳定的SEI膜来提高锂金属负极的稳定性[33-34,38]。同时由于高浓度电解液中有机溶剂的含量更少,其安全性能也能得到一定提高。例如,4 mol·L-1LiFSI溶解在DMC中形成的电解液,对比稀盐电解液具有明显更差的可燃性[20]。磷酸三乙酯(TEP)具有不易燃且抗氧化性能强的优点,但是其与锂金属负极兼容性差,通常只把它用作锂离子电池的阻燃添加剂,在LiFSI-TEP(摩尔比1∶2)高浓度电解液中,锂盐阴离子含量提高使该电解液不仅具有优质SEI膜形成能力,在Li‖Cu电池更呈现出高达99%的库仑效率,还具有优异的阻燃能力[50-51]。环丁砜(TMS)具有较强的抗氧化性能,但是TMS与锂金属的兼容性较差。高浓度策略能够有效提高TMS基电解液与锂金属的兼容性,LiFSI-3TMS电解液在Li‖Cu电池中CE高达98.8%,同时具有超过4.9 V的电化学稳定窗口[52]。氟代碳酸乙烯酯(FEC)是一种常用电解液添加剂,其具有高体积氟含量,在用于电解液添加剂时能够生成高氟含量的SEI膜。当直接使用FEC作高浓度LiFSI(7 mol·L-1)电解液的溶剂时,此电解液具有超过5 V的氧化电位,同时此电解液在锂金属表面形成高含氟的稳定SEI膜,这归功于FSI-与FEC两种电解液成分的高含氟量[29]。提高六氟磷酸锂基电解液浓度也能有效提高锂金属电池的CE[18]。
2.5 稠盐电解液稀释剂
虽然高浓度电解液具有众多优势,但是高浓度电解液通常也会带来多种负面影响。高浓度电解液通常具有更高的黏度,降低电解液对电极材料的润湿性,使得电池容量较低。过高的黏度也可能降低电解液的离子导电率,使电池的倍率性能变差,同时低导电率的电解液可能会使锂枝晶快速生长[6]。
为了保留高浓度电解液的优势,同时降低其黏度和提高离子导电率,人们找到一类只与溶剂互溶但不溶解锂盐的溶剂——稀释剂[52-56]。稀释剂的引入不仅明显降低了电解液的黏度,同时稀释剂的其他性能在电解液中得到保留。例如,环丁砜高浓度电解液中加入低熔点的1,1,2,2-四氟乙基-2,2,3,3-四氟丙基醚(TTE)能够极大降低电解液的黏度,提高电解液的离子导电率,同时TTE的应用使该电解液在-10 ℃下能够继续运行,极大地缓解了砜类高熔点的弊病[52]。LiFSI-FEC高浓度电解液中应用(四氟1-(2,2,2-三氟乙氧基)乙烷(D2)、甲氧基全氟丁烷(M3)等稀释剂赋予电解液-85~70 ℃超大温度区间的稳定循环性能,此类稀释剂的应用使得该电解液在全球所有极端自然温度下能够稳定运行[32]。碳酸二2,2,2-三氟乙基酯(TFEC)[57]、六氟异丙基甲基醚(HFME)[53]等稀释剂的应用极大降低了电解液可燃性。环己烷和正己烷用作电解液稀释剂能够有效降低锂金属形核过电位,改善锂金属沉积形貌[55]。同时,部分稀释剂同样能够帮助形成稳定的SEI膜,如三(三氟乙氧基)甲烷(TFEO)在LiFSI/DME电解液中能够帮助在锂金属表面形成非层状、非马赛克状的成分均匀的SEI膜[56]。
2.6 电解液添加剂

多种添加剂的联合使用在适当情况下能够进一步提高锂金属电池的性能。如在LiPF6/PC(碳酸丙烯酯)基电解液中加入VC,FEC与六氟砷酸锂(LiAsF6),能够明显改善锂沉积的形貌。当LiAsF6与VC或者FEC联合应用时具有最均匀的沉积形貌。VC与FEC具有相似的聚合成膜方式,但是FEC聚合时释放出氟化氢(HF)将进一步与锂金属发生反应。LiAsF6能够帮助在锂金属表面形成含有Li3As的合金层,此类型合金层有利于诱发锂形核并形成稳定SEI膜[69],同时LiAsF6分解有利于提高SEI膜中的LiF含量。SEI膜中高的LiF含量有利于降低锂离子扩散能垒,同时提高SEI膜的稳定性[11,66,70-71]。LiAsF6与VC或者FEC联合使用可产生高度致密、均匀、无枝晶以及自成簇的锂金属沉积层[72]。在0.6 mol·L-1LiTFSI-0.4 mol·L-1LiBOB/EC-EMC电解液中同时添加VC,FEC,LiPF6添加剂,对应Li‖Cu电池CE从91.9%提高到98.1%,明显高于单一添加剂。综上所述,通过操控添加剂实现了锂金属在碳酸酯类电解液中较高库仑效率的目的,产生均匀粗大结节状的无枝晶锂金属沉积形貌[73]。
使用添加剂能够在微小的添加量下明显地改变电池的循环性能,但是由于此类添加剂持续参与反应,在循环一段时间之后便可能失去其改善效果。
另外,并不是所有的添加剂都将参与反应。根据能斯特方程,如果一个阳离子(M+)其还原电位与Li+接近,那么(M+)在较低的浓度下的还原电位将可能低于Li+。例如,0.01 mol·L-1浓度下的Cs+有效还原电位(-3.144 V),具有比在1.0 mol·L-1浓度下的Li+(-3.040 V) 更低的有效还原电位。因此,当混合电解质中添加的Cs+的浓度明显低于Li+的浓度时,在Li+的沉积电位下Cs+将不会发生沉积,也不会在电极表面形成Li合金。在最初的沉积阶段,Li+与添加物Cs+都被吸附在电极(Li)表面。当外加电压(Va)略低于Li+离子的还原电位(ELi/Li+)但高于Cs+的还原电位(ECs/Cs+)时,即ELi/Li+>Va>ECs/Cs+,Li将沉积在电极表面。由于电极基板表面的不平整或者系统中的其他波动因素,一些Li凸起的尖端不可避免地会在底物表面形成。然后在新形成的Li凸起处出现更高的电子电荷密度,这将会吸引更多来自电解质的Li+并在尖端处沉积,这样在传统电解质中形成了Li枝晶。但是,大量的并不被还原的Cs+也将逐渐吸附到尖端形成正电屏蔽层并排斥锂离子,阻止其继续进入尖端沉积,因此锂离子只能在低洼处沉积,最终形成一个光滑平整的SEI膜[74]。硫脲小分子可被锂金属表面吸引,能够有效降低锂的形核功,促进形核,硫脲最易于被锂金属表面凹坑处吸引,因此将加速锂金属在凹坑处生长,最终形成均匀平整的沉积层[75]。此类非消耗类添加剂能够在电池持久运行中不失效。类似的还有二甲基乙酰胺[76]、丁二腈[77]、己二腈[78]等。己二腈能够作为一种双功能添加剂,在Li‖NCM电池中,通过帮助在锂负极产生更高LiF含量的SEI膜使锂沉积呈现光滑粗大的无枝晶形貌(LSV分析表明己二腈并不参与负极的反应),同时己二腈在正极活性材料表面与Ni4+强烈配位,极大降低了富Ni正极副反应的发生[78]。因其不直接参与反应,其改善效果持续时间更长。
一般来讲,目前先进的锂金属电池电解液Li‖Cu电池库仑效率应该在95%以上,表1统计了近年来通过电解液化学实现较高库仑效率的电解液配方及其应用于全电池的循环性能。
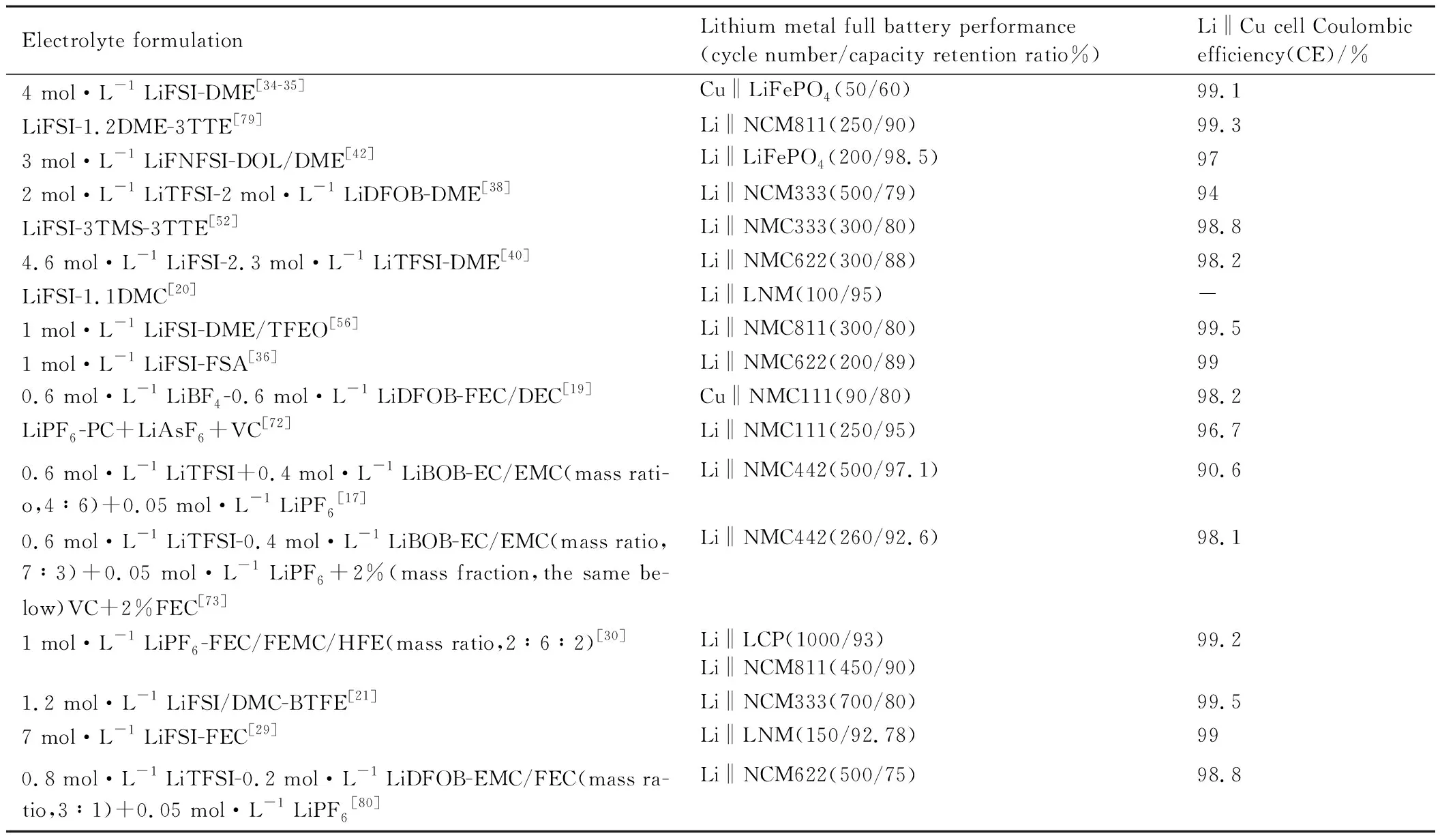
表1 电解液配方对应的锂金属全电池循环性能以及Li‖Cu电池库仑效率Table 1 Cycle performance of lithium metal full battery and Coulomb efficiency of Li‖Cu battery corresponding to electrolyte formulation
3 结束语
锂金属负极被认为是锂电池电极材料中的“圣杯”,综合性能最优异。但是由于锂金属的高活性特征,使其库仑效率低、易产生锂枝晶,严重影响了电池的电化学性能和安全性能,阻碍了其商业化进程。近年来,关于锂金属负极的研究已经取得较大进步,在锂枝晶形核和生长方面提出多种理论模型使人们逐渐深入理解锂枝晶的生长机理。并由此提出了多种抑制锂枝晶的方案,包括电解液化学、固态电解质、锂金属界面改性及锂金属微孔结构负极等。尽管如此,目前仍有很多问题亟待探索。锂枝晶的诸多生长模型,都限制在特定条件下,对其具体的形核、生长、微观结构等了解并不透彻,没有一个能够统筹说明其生长过程的理论。
操纵电解液化学是提升锂金属库仑效率、抑制锂枝晶生长的有效途径,且相对简便,实用性强。人们采用多种方法调控电解液化学,包括采用与锂金属兼容性更好的醚类溶剂、优质成膜锂盐、高浓度电解液、氟化溶剂、成膜添加剂以及调控锂金属界面双电层的添加剂等。醚类溶剂虽然是目前与锂金属兼容性最好的一种有机溶剂,但是较低的氧化电位和易燃的特性使其安全性能受到巨大挑战。虽然目前已经开发出几款具有较好成膜性能的锂盐,如LiDFOB,LiBOB等,但是LiDFOB在超过4.3 V易分解产生气体、LiBOB溶解度低等问题限制了其进一步应用。复合锂盐的使用在一定程度上能够使其优势互相补充,达到较好的综合效果。高浓度电解液策略能够有效提高锂金属的稳定性,形成更加稳定的SEI膜,但是高浓度同时也带来了过高的黏度以及高成本。使用合适的稀释剂能够使高浓度电解液保持高浓度的化学特征的同时降低电解液的黏度,但是目前对电解液稀释剂的研究大多数处于初级阶段,对其深层次化学机理的研究较少,在后续的研究中值得进一步深入。氟化溶剂通常使电解液具有更高的氧化电位,同时氟化溶剂中的F元素能够为SEI膜提供更多的F源,形成更高LiF含量的SEI膜。采用氟化溶剂配制锂金属电池电解液的研究目前处于起步阶段,在后续的研究中需要投入更多的关注,以促使高电压体系的锂金属电解液快速发展。添加剂的合理使用通常能够对锂金属电池的稳定性起到明显作用,调控双电层型添加剂能够吸附于电极表面来修饰SEI膜的性能,此类非消耗类添加剂能够在电池的整个运行过程中持续起到修饰效果。然而目前使用单一添加剂对锂金属的保护效果依然有限,适当将多种添加剂复合使用在某些情况下能够进一步提高锂金属的稳定性。
经过近10年的研究,锂金属电池的库仑效率从不到50%迅速提高到95%以上(目前报道中最高为99.5%[21,56]),为锂金属的实际应用提供了极大的信心。但是锂金属在多次沉积/剥离过程中“死”锂的形成以及锂金属与电解液间的副反应依然难以有效遏制,锂金属的库仑效率依然远未达到商业推广对库仑效率的底线要求(99.9%)。受限于目前的分析技术,液态电解质在锂金属界面产生SEI膜的形成机理、成分以及锂离子穿过SEI膜的确切过程和优质SEI膜可控性形成等仍然有待于进一步探索。适当利用溶剂、锂盐、添加剂三者间的协同效应,调控电解液本身的化学组成或者调控电极界面的双电层结构,降低电解液与锂金属间的反应性或者在锂金属表面生成锂离子通过效率更高的稳定的SEI膜,以促使更加均匀的锂沉积层的形成。此外,将电解液化学与锂金属多孔结构负极、锂金属界面修饰及固态电解质等多途径抑制锂枝晶的策略结合起来有望加速高能锂金属电池的商业化进程。
