ATP1A3基因突变致儿童快发病性肌张力障碍-帕金森综合征一例并文献复习
2021-07-06康庆云廖彩时廖红梅陈波杨理明
康庆云 廖彩时 廖红梅 陈波 杨理明
ATP1A3基因突变所致快发病性肌张力障碍-帕金森综合征(RDP)[1]是一种肌张力障碍叠加综合征,属国际肌张力障碍分类12型,其临床特征鲜明,以数小时至30天快速发病的肌张力障碍和帕金森综合征为特征[2]。ATP1A3基因突变所致RDP是一种临床极为罕见的运动障碍性疾病,目前世界范围内报道的病例仅61例,国内至今仅3篇文献共6例患者被报道[3-5]。本文回顾分析1例儿童期发病、经基因检测确诊的ATP1A3基因突变所致RDP患儿的临床资料,通过复习相关文献总结该综合征特点,以期提高临床医师对该病的认识水平。
病例资料
患儿 女性,4岁9个月。因发热4天伴乏力、意识障碍3天,于2019年3月6日入院。患儿4天前因“受凉”而诱发发热、头痛及咽痛,次日晨起出现肢体乏力、竖头不稳、不能直立、嗜睡等症状与体征,遂至当地医院就诊,腰椎穿刺脑脊液及头部MRI检查均未发现明显异常,考虑“脑干脑炎”,予抗病毒中药炎琥宁(80 mg/d)和多种维生素对症支持治疗,连续治疗3天症状无缓解且逐渐出现吞咽困难、失语等症状,为求进一步诊断与治疗至我院就诊,以“颅内感染”原因待查收入院。自发病以来呈嗜睡状态,大小便正常,体重无明显变化。
既往史、个人史及家族史 系孕2产2、足月顺产,智力、运动发育里程碑正常,3月龄抬头、7月龄独坐、12月龄独走。既往体格健康,发病前可自行进食、如厕,与他人交流无语言障碍。父母体格健康,无同类疾病或家族遗传性疾病病史。
入院后体格检查 患儿体温为36.5℃,心率为102次/min,呼吸为21次/min,血压为130/80 mm Hg(1 mm Hg=0.133 kPa);一般内科检查无异常。神经系统检查呈嗜睡状态,哭闹不安,无言语交流;颈软;眼睑无下垂、无眼震,眼球活动正常;口角无歪斜,咽反射减弱;竖头不稳、独坐不能,粗测双上肢肌力2级、双下肢肌力3级,四肢肌张力低下;双侧膝反射正常,跟腱反射正常,Kernig征、Brudzinski征均呈阴性,左侧Babinski征阳性、右侧为阴性。
辅助检查 实验室检查血尿便常规、肝肾功能、心肌酶谱、电解质及血糖等项指标均于正常值范围;乳酸、铜蓝蛋白、血尿串联质谱分析、脑脊液检查无异常;血、脑脊液自身免疫性脑炎抗体呈阴性。脑电图背景节律慢化,肌电图无异常。头部MRI(平扫和增强)、fMRI、脊髓MRI(平扫和增强),以及胸部X线检查均无异常所见,眼底、腹部彩超检查正常。入院15天(2019年3月21日)行图片词汇测试,原始总评分为10,百分位数<1,提示存在认识功能减退可能。考虑患儿肌张力障碍累及部位有明显的头腿梯度差(面部>上肢>下肢),为明确诊断,经患儿父母知情同意于入院22天(2019年3月28日)采集患儿及其父母外周静脉血2 ml,采用外显子芯片捕获+高通量测序技术进行第二代基因组DNA全外显子组测序(WES,北京金准基因科技有限责任公司),根据测序结果设计聚合酶链反应(PCR)扩增引物,采用Sanger测序并验证。结果显示,患儿存在ATP1A3基因c.2267G>A(p.R756H)位点杂合错义突变,家系验证其父母均未携带同型杂合突变,为新生突变(图1)。最终确诊为RDP。
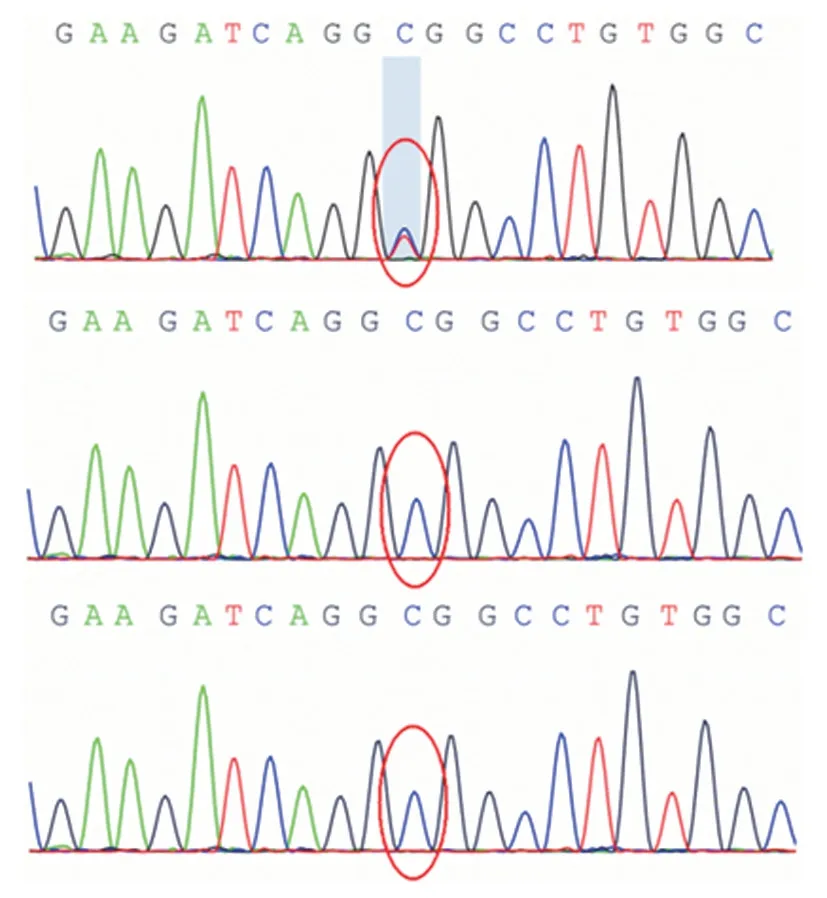
图1 基因检测结果 1a 先证者存在ATP1A3基因c.2267G>A(p.R756H)位点杂合错义突变(红圈所示) 1b,1c 先证者父母不存在ATP1A3基因c.2267G>A(p.R756H)位点突变(红圈所示)Figure1 Genetictestingfindings Theprobandhad heterozygositymissensemutationinthec.2267G>A(p.R756H)siteofATP1A3 gene(red circleindicates,Panel1a).Theproband'sfatherandmotherhadno mutationatthissite(redcirclesindicate;Panel1b,1c).
患儿入院以后,经体格检查和各项辅助检查考虑为脑干脑炎或吉兰-巴雷综合征(GBS)谱系疾病,治疗原则以抗炎、调节免疫为主。予以静脉注射免疫球蛋白400 mg/kg(1次/d×5 d)和奥拉西坦1 g/d(1次/d×10 d)静脉滴注,同时辅助神经营养药、康复综合训练,连续治疗12天(2019年3月18日)患儿神志逐渐清醒,可自主少量进食但流涎症状明显,仍无明确的语言交流,肢体肌力有所改善(双上肢肌力3级、双下肢肌力4级)但竖头不稳、独坐不能如前;入院19天(2019年3月25日)时出现不自主甩头动作,同时伴有肢体及躯干舞蹈徐动样动作,此时肢体肌力虽已明显好转,但竖头及独坐不稳症状仍未缓解,建议予以氯硝西泮治疗,但未获得患儿父母同意。2019年5月6日经基因检测确诊为RDP,开始规律服用氯硝西泮0.10mg/kg(2次/d),连续治疗26天(2019年6月1日)后可独坐;72天(2019年7月17日)可独自行走数步,言语交流呈单词式且吐词不清;2.50个月(2019年7月20日)可独自行走但呈宽基底步态;4个月(2019年9月4日)能自主持勺进食;5个月(2019年10月15日)可双足跳,自主穿、脱衣但协调性稍差,面部表情欠丰富,可说短句,发音略含糊。末次随访时(2019年11月30日)肢体肌力基本如常,但协调性稍差,语言表达欠流畅,构音障碍。
讨 论
笔 者 以“RDP”、“Rapid-onsetdystonia parkinsonism”、“DYT-12”、“快发病性肌张力障碍-帕金森综合征”等词组作为关键词,分别检索美国国立医学图书馆生物医学文献数据库(PubMed)、万方数据知识服务平台、中国知网中国知识基础设施工程(CNKI)等数据库(建库至2019年12月),共获得RDP相关文献126篇61例病例,国外报道55例、国内报道6例(表1)。
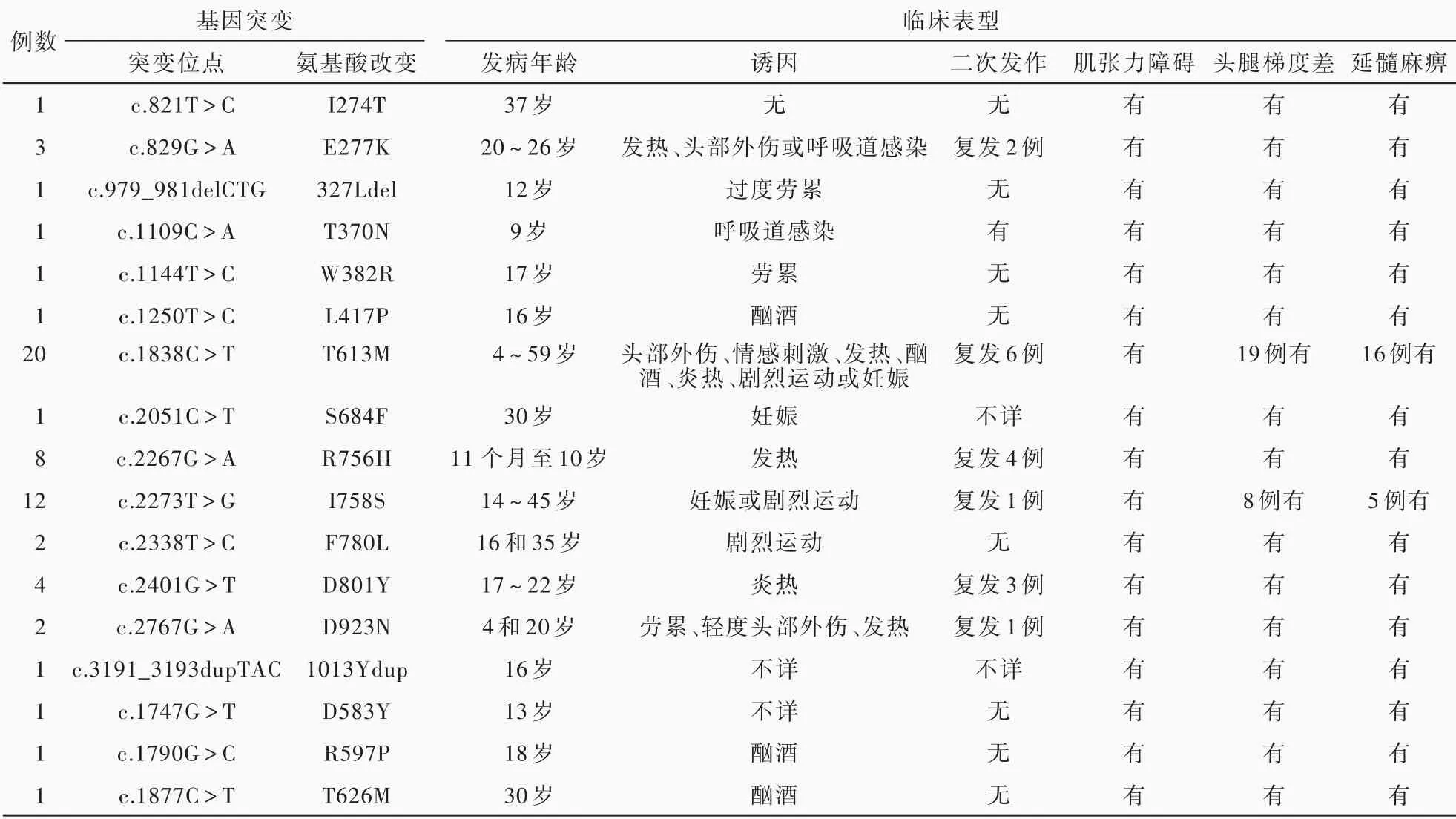
表1 文献报道的61例RDP患者临床资料Table 1. Clinical data of 61 patients with RDP reported in the literature
RDP由Dobyns等[6]于1993年首次报告,以肌张力障碍为主要表现,多于青少年期发病,感冒、妊娠、酗酒、情感打击等为常见诱发原因[7];典型症状表现为突发性肢体肌张力障碍伴随运动迟缓、姿势不稳、吞咽困难,以及进行性构音障碍等[8]。诊断标准包括[9]:(1)数分钟至30天内突发快速进展的肌张力障碍和帕金森样症状。(2)肌张力障碍累及部位且具有明显的头腿梯度差(面部>上肢>下肢)。(3)呈明显的球部受累表现。(4)对左旋多巴治疗不敏感。(5)符合常染色体显性遗传家族史或新生突变。本文患儿以“感染”为诱发因素,伴随急性快速进展的肌张力障碍,受累部位具有明显的头腿梯度差,病程中表现为吞咽困难、失语等延髓受累症状,并逐渐出现舞蹈样动作、姿势不稳等帕金森样症状,上述症状于发病1个月后趋于稳定,符合RDP诊断。幻觉、抑郁、焦虑等精神症状和认知功能减退等非运动症状在RDP患者中并不少见[10-11],本文患儿图片词汇测试结果提示存在认识功能减退。
ATP1A3基因于2000年被Pittock等[1]确定为RDP的致病基因,deCarvalhoAguiar等[12]于2004年首次克隆ATP1A3基因,可编码钠-钾ATP泵上的α3亚基,在小脑、基底节、海马、丘脑等区域神经元中呈高表达,具有维持细胞内外钠、钾离子交换、保证神经元电兴奋性和神经递质跨膜转运等重要生理功能[6,13]。目前已报道的ATP1A3基因突变类型共有17种,以T613M突变位点最为常见[2,10],其次是E277K和R756H突变位点[2,14]。本文患儿存在ATP1A3基因c.2267G>A(p.R756H)位点杂合错义突变,为RDP致病性突变,支持RDP诊断。ATP1A3基因存在外显率不全现象[15],故部分家系病例症状不典型,甚至有的基因突变患者完全无症状,某些基因位点突变可引起儿童交替性偏瘫(AHC)和(或)RDP中间型,这在D583Y、G867D、E951K、D801N及R756C位点突变中均有报道[16-18]。
除RDP外,ATP1A3基因突变与多种中枢神经系统疾病有关,如交替性偏瘫、小脑共济失调、腱反射消失、高足弓、视神经萎缩、感觉神经性耳聋综合征、婴儿早期癫性脑病和复发性脑病伴小脑共济失调等[9,19-22]。不同神经系统疾病ATP1A3基因突变的位点有所不同,交替性偏瘫最为常见的3种基因突变位点分别为E815K、D801N以及G947R[23],感觉神经性耳聋综合征目前仅有E818K一种突变位点[22,24-25],而复发性脑病伴小脑共济失调则均为第756位精氨酸变异[26]。同一基因位点突变亦可引起不同的临床表型,D923N和E277K突变在RDP和交替性偏瘫患者中均有报道[27];RDP及复发性脑病伴小脑共济失调均存在R756H突变,好发于女性病例,肌力和肌张力下降表现更为突出[21],与本文患儿临床特征相吻合。
本文患儿存在R756H突变,RDP临床症状典型,目前与该患儿具有相同突变位点的RDP病例共报道8例[3,5,14,28-29],均于儿童期发病,表现为由发热诱发的构音困难、肌无力同时伴有帕金森样症状。其中,2例婴儿期发病者表现为RDP重叠交替性偏瘫[14,28],在病程中有二次发作,考虑临床表型为婴儿变异型RDP;另有3例患者来自同一家系[2],均具有幼儿期感染诱发后快速发病的特点,其中先证者病程中有二次发作,其兄及母均为典型的RDP临床表型。既往文献报道RDP儿童期发病罕见,且罕有二次发作[20],而包括本文患儿在内的9例ATP1A3基因R756H突变患者均于儿童期发病[3,5,14,28-29],且有4例患者病程中有二次或多次发作[3,5,14,28],提示儿童期发病的RDP可能更倾向于多次发作,并常与交替性偏瘫临床表型重叠。
目前针对RDP尚无确切有效的治疗方法。根据文献报道,大部分RDP患者对多巴胺受体激动药(普拉克索)、拟多巴胺药(左旋多巴)、中枢抗胆碱药、氟桂利嗪或巴氯芬等治疗不敏感[7,13];部分患者大剂量苯二氮类药物可获得部分缓解[1-2,13];对药物治疗效果差的患者,可尝试行脑深部电刺激术(DBS)治疗[28],但亦有报道称DBS治疗RDP是无效的[30]。另外,可根据RDP患者的临床症状进行包括吞咽训练和康复综合治疗在内的对症支持治疗,对合并抑郁、焦虑等精神症状者给予相应药物对症治疗,可取得一定效果。本文患儿经氯硝基安定等药物治疗,同时辅助吞咽功能训练及康复综合治疗,症状明显改善。
RDP临床特征鲜明,急性发病,不明原因的运动障碍,伴明显的构音障碍、吞咽障碍等延髓受累体征,且运动障碍有明确的头腿梯度差特点而头部MRI等各项检查无明显异常,此类患者应考虑RDP的可能,及时行ATP1A3基因检测将有助于早期诊断与治疗,以及优生优育。
利益冲突 无
