过氧化物酶体酰基辅酶A氧化酶缺乏症一例并文献复习
2021-07-06尤桦菁田杨李洵桦李小晶裴中
尤桦菁 田杨 李洵桦 李小晶 裴中
过氧化物酶体酰基辅酶A氧化酶缺乏症[在线人类孟德尔遗传数据库(OMIM#264470)]系第17号染色体ACOX1基因突变导致的常染色体隐性遗传性单一酶缺乏症,最早由Poll-The等[1]于1988年报告。过氧化物酶体参与细胞内极长链脂肪酸(VLCFAs)的β-氧化以及胆汁酸和植烷酸等磷脂的合成,其功能障碍可以导致新生儿肾上腺脑白质营养不良(NALD)、Zellweger综合征(ZS)和婴儿型Refsum综合征(IRS)等多种过氧化物酶体病[2]。极长链脂肪酸的β-氧化通路先后由酰基辅酶A氧化酶、包含烯酰辅酶A水合酶和3-羟基酰基辅酶A脱氢酶的双功能酶、3-酮脂酰辅酶A硫解酶催化而成,过氧化物酶体酰基辅酶A氧化酶缺乏症是该通路的首个催化酶酰基辅酶A氧化酶功能障碍所致[1](图1)。国外已报道34例患者[1-13],但目前国内尚无相关报道。中山大学附属第一医院神经科和广东省广州市妇女儿童医疗中心神经内科共同诊断与治疗1例ACOX1基因突变致过氧化物酶体酰基辅酶A氧化酶缺乏症患儿,并结合文献综述其临床特征,以期提高临床医师对该病的认识。

图1 极长链脂肪酸的β-氧化通路以及该通路上各种催化酶缺乏导致的过氧化物酶体病Figure 1 β-oxidation pathway of VLCAFs and peroxisomal disorders caused by various enzymatic deficiency on this pathway.
病例资料
患儿 男性,3岁,主因自幼发育迟缓,行走障碍1月余,于2020年3月13日收入广东省广州市妇女儿童医疗中心。患儿胎龄39周时顺产,产程中无窒息史和产伤史,出生体重不详,出生时肌张力偏低,因“新生儿吸入综合征、新生儿轻度窒息及心肌损害”在当地医院住院治疗;父母诉其自幼发育迟缓,长期至当地医院儿童保健所随诊,患儿6个月可抬头、10个月可独坐、12个月可爬行、17个月可独自站立和行走、18个月可说单字或简单词语。5月龄时曾发生1次抽搐发作(具体不详),当时体温<38.5℃,当地医院头部CT和脑电图检查未见明显异常,此后未再发作。1个月前(2020年2月)无明显诱因开始出现行走不稳,步态较宽,伴右侧跛行,同时出现头部不自主右偏,伴流涎,偶有饮水呛咳,可进食固体食物,易激惹,精神反应欠佳;当地医院头部MRI检查(2020年2月18日)显示,脑桥和双侧小脑半球异常信号影;临床疑诊“脑炎”,静脉注射地塞米松2.50 mg连续5天、间隔3天,再静脉注射地塞米松2.50 mg连续4天和免疫球蛋白2.50 g仅1次,症状无明显好转,右下肢跛行症状逐渐加重至无法独坐、独自站立和行走,并出现右上肢无力,持物无力以左手显著,伴双手伸向物体时出现震颤,语言表达能力倒退,发病前可表达句子、发病后单字延长且以单字为主。
入院后体格检查 神志清楚,言语不清且以单字为主,前额突起,低鼻梁,反应淡漠,双侧眼动欠灵活、未见眼震,四肢肌力3~4级、肌张力增高,尤以双下肢显著,痉挛步态,指鼻试验、跟-膝-胫试验和Romberg征不配合,意向性震颤可疑阳性,感觉系统查体不配合,四肢腱反射亢进,病理反射未引出,脑膜刺激征阴性。
辅助检查 动脉血气分析、血浆氨基酸、血浆酰基肉碱、血清氨、血清酮体/乳酸丙酮酸比值均于正常值范围,腰椎穿刺脑脊液常规、生化、病原体、副肿瘤综合征(PNS)抗体、中枢神经系统脱髓鞘抗体、神经鞘脂贮积病酶学测定等亦于正常值范围,尿代谢物气相色谱-串联质谱(GC-MS/MS)检测、腹部超声和脑电图未见异常。头部MRI检查(2020年3月17日)显示,延髓、中脑腹侧、脑桥、双侧小脑齿状核和颈髓可见对称性片状异常信号影,T1WI呈稍低信号、T2WI呈高信号(图2)。MRS显示,双侧小脑齿状核胆碱(Cho)峰和乳酸(Lac)峰升高,N-乙酰天冬氨酸(NAA)峰下降(图3)。脑干听觉诱发电位(BAEP,2020年3月20日)监测显示,双侧听觉传导通路损害,双侧Ⅲ波和Ⅴ波潜伏期延长,Ⅰ~Ⅲ波间期和Ⅰ~Ⅴ波间期延长;视觉诱发电位(VEP)监测显示,双侧P100潜伏期延长。复查头部和颈椎MRI(2020年3月27日)显示,延髓、中脑腹侧、脑桥、双侧小脑齿状核和颈髓对称性片状异常信号影大致同前,考虑代谢性脑病可能(图4,5)。进一步行血清VLCFAs测定(2020年4月21日),其结果显示,C22:0值 为105μmol/L(<104.27μmol/L)、C24:0 94.50μmol/L(<104.29μmol/L)、C26:0 2.56μmol/L(<0.89μmol/L),C24:0/C22:0比值为0.90(<1.04)、C26:0/C22:0比值为0.024(<0.013)。

图2 头部MRI检查(2020年3月17日)所见 2a 横断面T1WI显示,中脑腹侧对称性片状稍低信号影(箭头所示) 2b 横断面T2WI显示,中脑腹侧对称性片状高信号影(箭头所示) 2c 横断面抑脂FLAIR成像显示,中脑腹侧对称性片状高信号影(箭头所示) 2d 横断面T1WI显示,脑桥和双侧小脑齿状核对称性片状稍低信号影(箭头所示) 2e 横断面T2WI显示,脑桥和双侧小脑齿状核对称性片状高信号影(箭头所示) 2f 横断面抑脂FLAIR成像显示,脑桥和双侧小脑齿状核对称性片状高信号影(箭头所示)Figure2 CranialMRIfindingsonMarch17,2020 AxialT1WIshowedsymmetricallypatchyand slighthypointensity in ventralmidbrain (arrow indicates,Panel2a). AxialT2WI showed symmetricallypatchyhyperintensityinventralmidbrain(arrow indicates,Panel2b). Axialfat suppressionFLAIRshowedsymmetricallypatchyhyperintensityinventralmidbrain(arrowindicates,Panel2c).AxialT1WIshowedsymmetricallypatchyandslighthypointensityinponsandbilateral cerebellardenatenuclei(arrow indicates,Panel2d). AxialT2WIshowedsymmetricallypatchy hyperintensityinponsandbilateralcerebellardenatenuclei(arrowindicates,Panel2e).Axialfat suppressionFLAIR showed symmetricallypatchyhyperintensityinponsandbilateralcerebellar denatenuclei(arrowindicates,Panel2f).

图4 复查头部MRI(2020年3月27日)所见 4a 横断面T1WI显示,中脑腹侧对称性片状稍低信号影(箭头所示),较前无明显变化 4b 横断面T2WI显示,中脑腹侧对称性片状高信号影(箭头所示),较前无明显变化 4c 横断面FLAIR成像显示,中脑腹侧对称性片状高信号影(箭头所示),较前无明显变化 4d 横断面T1WI显示,脑桥和双侧小脑齿状核对称性片状稍低信号影(箭头所示),较前无明显变化 4e 横断面T2WI显示,脑桥和双侧小脑齿状核对称性片状高信号影(箭头所示),较前无明显变化 4f 横断面FLAIR成像显示,脑桥和双侧小脑齿状核对称性片状高信号影(箭头所示),较前无明显变化Figure 4 Cranial MRI findings on March 27,2020 Axial T1WI showed symmetrically patchy and slight hypointensity in ventral midbrain(arrow indicates,Panel 4a),which was similar to the previous T1WI.Axial T2WI showed symmetrically patchy hyperintensity in ventral midbrain(arrow indicates,Panel 4b),which was similar to the previous T2WI.Axial FLAIR showed symmetrically patchy hyperintensity in ventral midbrain(arrow indicates,Panel 4c),which was similar to the previous FLAIR.Axial T1WI showed symmetrically patchy and slight hypointensity in pons and bilateral cerebellar denate nuclei(arrow indicates,Panel 4d),which was similar to the previous T1WI.Axial T2WI showed symmetrically patchy hyperintensity in pons and bilateral cerebellar denate nuclei(arrow indicates,Panel 4e),which was similar to the previous T2WI. Axial FLAIR showed symmetrically patchy hyperintensity in pons and bilateral cerebellar denate nuclei(arrow indicates,Panel 4f),which was similar to the previous FLAIR.
基因检测 进一步追问个人史及家族史,患儿出生后混合喂养,按时添加辅食;父母为四代近亲婚配,其兄出生时尿道下裂,手术治愈,但无类似症状。遂于2020年4月14日抽取患儿及其父母外周静脉血各2 ml,送检广州嘉检医学检测有限公司行全外显子组测序(WES),结果显示,患儿存在ACOX1基因c.1589A>G(p.His530Arg)纯合突变,其父母均携带ACOX1基因c.1589A>G(p.His530Arg)杂合突变(图6),但无临床症状。该位点在过氧化物酶体酰基辅酶A氧化酶缺乏症患者中暂无报道,亦未在ClinVar数据库(www.ncbi.nlm.nih.gov/clinvar/ClinVar)中检索到。结合患儿临床表现、影像学显示的双侧脑白质多发性对称病灶、血清VLCFAs升高,及该变异所在氨基酸区域高度保守,SIFT软件(http://provean.jcvi.org/index.php)预测为“影响蛋白质功能可能性大”,PolyPhen-2软件(http://genetics.bwh.harvard.edu/pph2/index.shtml)预 测 为“可 能 致病”;其父母近亲婚配且为该变异携带者,根据美国医学遗传学和基因组学会(ACMG)指南[14],认为该变异为可能致病突变。为明确该变异的致病性,建议进一步行酶的活性测定或蛋白质功能试验。
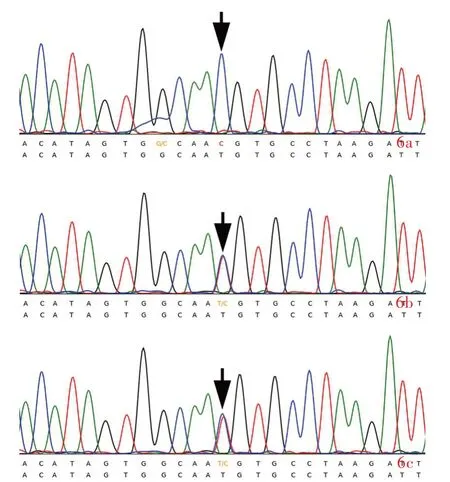
图6 全外显子组测序所见 6a 患儿存在ACOX1基因c.1589A>G(p.His530Arg)纯合突变(箭头所示)6b 患儿之父携带ACOX1基因c.1589A>G(p.His530Arg)杂合突变(箭头所示) 6c 患儿之母携带ACOX1基因c.1589A>G(p.His530Arg)杂合突变(箭头所示)Figur e 6 Whole exome sequencing findings ACOX1 gene c.1589A>G(p.His530Arg)homozygous mutation(arrow indicates)in the patient(Panel 6a).ACOX1 gene c.1589A>G(p.His530Arg)hererozygous mutation(arrow indicates)carried by his father(Panel 6b).ACOX1 gene c.1589A>G(p.His530Arg)hererozygous mutation(arrow indicates)carried by his mother(Panel 6c).
诊断与治疗 结合患儿临床表现、影像学和实验室检查、基因检测,最终经中山大学附属第一医院神经科明确诊断为过氧化物酶体酰基辅酶A氧化酶缺乏症。住院期间予甲泼尼龙25 mg/d静脉滴注连续7天后改为20 mg/d静脉滴注1天,及静脉注射免疫球蛋白7.50和5 g/d各1天,同时予辅酶Q10(能气朗)10 mg/d(3次/d)口服连续7天后改为20 mg/d(3次/d)连续10天、左卡尼汀1 g/d静脉滴注连续治疗9天,复合维生素B口服11天和维生素B220 mg/d(3次/d)口服连续2天等对症治疗。共住院16天,出院时精神稍好转,可独坐和独自站立数秒,头可竖立,但与父母无语言沟通,偶有口角流涎和饮水呛咳,查体基本同前。患儿预后较差,已失访。
讨 论
过氧化物酶体酰基辅酶A氧化酶缺乏症临床主要表现为新生儿期肌张力低下、癫发作和生长发育迟缓,同时可出现感觉神经性耳聋、色素性视网膜炎、肝肿大和特殊面容等临床特征(表1)。癫发作形式和发病年龄具有较高的临床异质性,发作形式主要包括局灶性发作和全面性强直-阵挛发作(GTCS)等,其发病年龄为新生儿期至6岁[3]。出现神经功能退化前,患儿一般可独坐、独自行走,可表达单字、单词或短句[4];大多数患儿于2~4岁出现神经功能退化现象,亦有文献报道2例过氧化物酶体酰基辅酶A氧化酶缺乏症患儿于于8~10岁发病,至50余岁时神经功能损害仍不严重[5];患儿通常于4~10岁死亡,除上述文献报道的2例发病较晚且进展缓慢的患者外,Suzuki等[6]还报告1例生存至19岁的患者。

表1 既往文献报道和本文患儿的临床特征Table 1. Clinical characteristics of our child patient and those in previous literature
过氧化物酶体酰基辅酶A氧化酶缺乏症的临床症状与新生儿肾上腺脑白质营养不良和Zellweger综合征等过氧化物酶体病相似,均可表现为新生儿期发病的肌张力低下、癫发作、生长发育迟缓和特殊面容等。过氧化物酶体酰基辅酶A氧化酶缺乏症最早由Poll-The等[1]于1988年报告,2例患儿误诊为新生儿肾上腺脑白质营养不良,行肝脏组织穿刺活检后发现过氧化物酶体含量不变但体积增大,更重要的是过氧化物酶体内的酰基辅酶A氧化酶缺乏。此后,Watkins等[11]发现,过氧化物酶体酰基辅酶A氧化酶缺乏症患儿神经功能退化的年龄相对较晚且生存期相对较长,而新生儿肾上腺脑白质营养不良和Zellweger综合征患儿通常1岁内即出现病情恶化并死亡。根据其发病机制,新生儿肾上腺脑白质营养不良和Zellweger综合征均属于过氧化物酶体合成障碍性疾病,其中Zellweger综合征患儿过氧化物酶体以及酰基辅酶A氧化酶、双功能酶(烯酰辅酶A水合酶和3-羟基酰基辅酶A脱氢酶)、3-酮脂酰辅酶A硫解酶这3种催化酶含量急剧下降,新生儿肾上腺脑白质营养不良患儿过氧化物酶体含量部分下降且双功能酶缺乏;而过氧化物酶体酰基辅酶A氧化酶缺乏症属于单一酶缺乏病,因此,3种疾病的病情严重程度依次为Zellweger综合征、新生儿肾上腺脑白质营养不良和过氧化物酶体酰基辅酶A氧化酶缺乏症[12]。
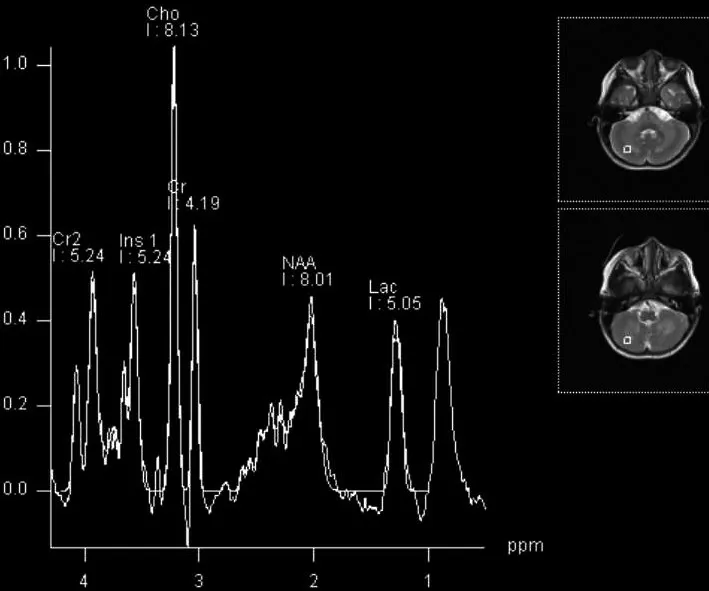
Cr,肌酐;Ins,肌醇;Cho,胆碱;NAA,N-乙酰天冬氨酸;Lac,乳酸图3 MRS显示,小脑齿状核Cho峰升高Figure 3 MRS showed elevated Cho peak in cerebellar dentate nucleus.
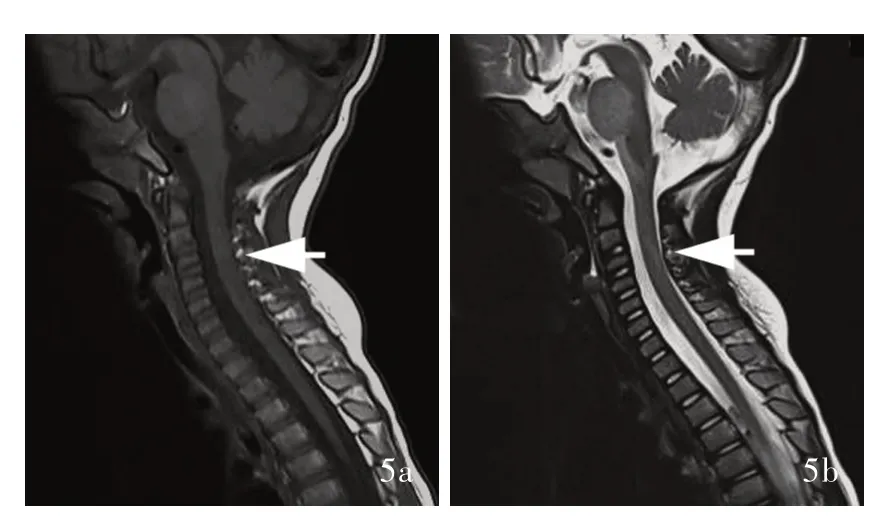
图5 复查颈椎MRI(2020年3月27日)所见 5a 矢状位T1WI显示,延髓、中脑腹侧、脑桥和颈髓稍低信号影(箭头所示) 5b 矢状位T2WI显示,延髓、中脑腹侧、脑桥和颈髓高信号影(箭头所示)Figure 5 Recheck cervical MRI findings on March 27,2020 Sagittal T1WI showed slight hypointensity in ventral midbrain, pons and cervical cord (arrow indicates, Panel 5a). Sagittal T2WI showed hyperintensity in ventral midbrain,pons and cervical cord(arrow indicates,Panel 5b).
过氧化物酶体酰基辅酶A氧化酶缺乏症的MRI可见小脑、脑干、大脑皮质下白质对称性炎性脱髓鞘性病变,T1WI呈低信号、T2WI呈高信号,且与临床严重程度相关[13]。神经功能退化前,MRI可能无明显病灶,VLCFAs累积被认为是病程进展后脑白质脱髓鞘的主要原因[15]。MRI异常改变通常先见于小脑白质、小脑脚和脑干,伴随小脑蚓部萎缩,随着病程进展,脑干上部锥体束、内囊后肢、侧脑室周围白质和胼胝体压部相继出现异常改变,逐渐蔓延至额叶白质[5,16]。因此,小脑白质和脑干脱髓鞘改变可能是过氧化物酶体酰基辅酶A氧化酶缺乏症患儿神经功能退化的早期特征,故MRI可作为预后判断的重要检查方法[17]。本文患儿出现行走不稳等症状后,首次行头部MRI检查显示脑桥和双侧小脑半球异常信号,1个月后上述症状加重并复查MRI,腹侧中脑、脑桥、延髓和双侧小脑齿状核均可见异常信号,提示病程进展较快且预后不良。
酰基辅酶A是相对分子质量为34×103的一种催化酶,其羧基末端(C端)氨基酸序列与大鼠具有高达88%的同源性,Ser-Lys-Leu残基作为过氧化物酶体靶向信号可介导基质蛋白进入并形成成熟的过氧化物酶体[2]。Watkins等[11]采用免疫组化和超微结构观察,发现过氧化物酶体酰基辅酶A氧化酶缺乏症患儿组织细胞中过氧化物酶体体积增大但含量保持不变。Ferdinandusse等[4]的研究显示,过氧化物酶体酰基辅酶A氧化酶缺乏症患儿的血浆和纤维母细胞中过氧化物酶体β-氧化和酰基辅酶A活性均低于正常对照者,致使C24:0和C26:0在体内累积。本文患儿存在视觉通路损害,但未配合行眼科检查。文献报道,多数过氧化物酶体酰基辅酶A氧化酶缺乏症患儿存在视网膜病变,其中二十二碳六烯酸(DHA,C22:6n-3)是哺乳动物视网膜中最为重要的多不饱和脂肪酸(PUFA),其生物合成通路包括内质网和过氧化物酶体,饮食中摄入的亚麻酸(C18:3n-3)在内质网中经一系列延长和去饱和作用形成C24:6n-3,再在过氧化物酶体中将C24:6n-3经β-氧化为C22:6n-3[7-8]。因此,当过氧化物酶体缺乏酰基辅酶A氧化酶时,无法正常合成C22:6n-3,从而导致视网膜病变。过氧化物酶体酰基辅酶A氧化酶缺乏症的突变基因最早由Fournier等[2]于1994年经cDNA克隆所发现,目前已有多篇文献报道ACOX1基因错义突变、截短突变、无义突变等多种突变类型,但临床表型与基因型并无关联性,临床症状严重程度与血浆VLCFAs和β-氧化活性之间亦无明显关联性[3-4]。另外,患儿的父母及兄长无类似症状,父母已被验证为携带者,若其兄也行同一突变位点验证,可进一步证实该变异的致病性。
目前尚无特异性有效治疗方法。2014年,Wang等[9]报告1例3岁男性过氧化物酶体酰基辅酶A氧化酶缺乏症患儿,予以造血干细胞移植(HSCT)治疗,其姐同为过氧化物酶体酰基辅酶A氧化酶缺乏症患儿,作为对照未行造血干细胞移植治疗,结果显示,该疗法并未阻止过氧化物酶体酰基辅酶A氧化酶缺乏症的病程进展,但脑组织尸检显示,患儿大脑皮质萎缩、神经元凋亡和神经炎症等均较其姐有所改善。2017年,Eichler等[10]报告造血干细胞基因治疗X-连锁肾上腺脑白质营养不良的Ⅱ和Ⅲ期临床试验结果,通过向患者输注在体外被包含ABCD1 cDNA的Lenti-D慢病毒载体转导的自体CD34+造血干细胞,发现所有患者均可检测到肾上腺脑白质发育不良蛋白(ALDPs),绝大部分患者生存且临床症状轻微。总之,过氧化物酶体酰基辅酶A氧化酶缺乏症、新生儿肾上腺脑白质营养不良和Zellweger综合征均为单基因突变导致的过氧化物酶体病,过氧化物酶体含量和(或)催化酶均有不同程度减少,目前造血干细胞移植已用于治疗过氧化物酶体酰基辅酶A氧化酶缺乏症,提示造血干细胞基因治疗也可能适用于过氧化物酶体酰基辅酶A氧化酶缺乏症及其他过氧化物酶体病(表2)。临床实践中,应提高患儿家属对疾病的认识并进行生育指导,如禁止近亲婚配、建议妊娠期女性携带者进行产前诊断等[18-19]。
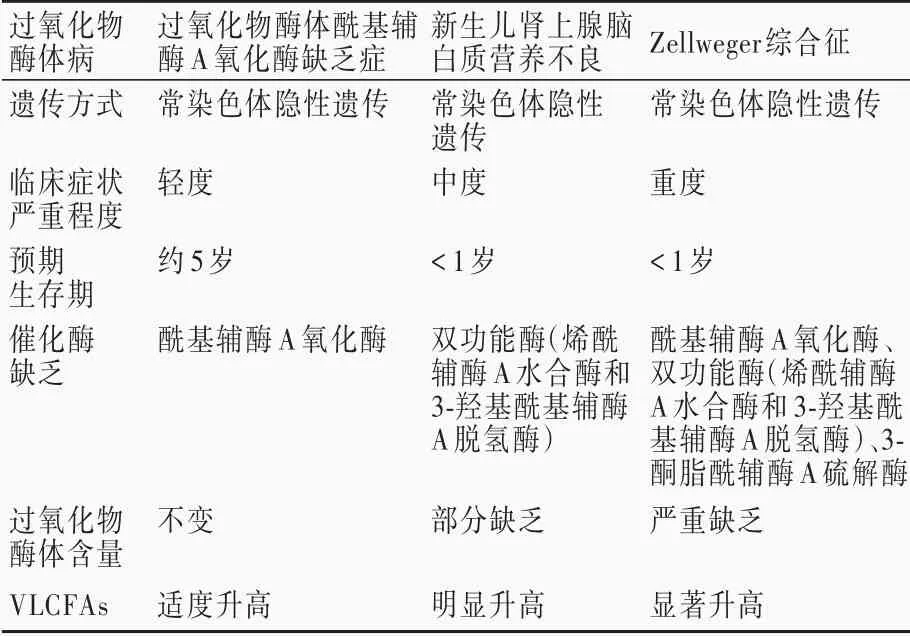
表2 过氧化物酶体病的临床特征及发病机制Table 2. Clinical characteristics and comparison of pathogenesis of various peroxisomal disorders
综上所述,本文首次报道ACOX1基因c.1589A>G(p.His530Arg)突变,但仍需进一步行蛋白质功能试验以验证该变异的致病性。对于新生儿期发病的肌张力低下、癫发作和生长发育迟缓,若头部MRI显示小脑、脑干等对称性异常信号,应高度警惕过氧化物酶体病,阳性家族史、VLCFAs水平升高和基因检测可明确诊断。过氧化物酶体酰基辅酶A氧化酶缺乏症目前尚无有效治疗方法,尽早确诊有助于对患儿及其家庭进行宣传教育、遗传咨询和产前诊断。
利益冲突 无
