绵马贯众有效部位微丸的处方工艺优化及溶出特性研究
2021-07-03杨锦高增平尹兴斌曲昌海倪健北京中医药大学中药学院北京102488
杨锦,高增平,尹兴斌,曲昌海,倪健(北京中医药大学中药学院,北京 102488)
绵马贯众为鳞毛蕨科植物粗茎鳞毛蕨Dryopteris crassirhizomaNakai的干燥根茎和叶柄残基,味苦,性微寒,有小毒,归肝、胃经,具有清热解毒、驱虫的功效,常用于治疗时疫感冒、风热头痛、温毒发斑、疮疡肿毒、崩漏下血、虫积腹痛。现代研究中发现多个含有绵马贯众的中药复方被用于治疗流感,如双贯清感滴丸、连花清瘟胶囊和贯黄感冒颗粒等[1-4]。
绵马贯众中的有效成分绵马贯众素ABBA具有良好的抗流感病毒作用[5],对感染流感病毒小鼠的保护率达90%。本课题组采用特殊提取工艺,对其有效成分进行富集,成功制得绵马贯众有效部位(GZP),主成分含量超过50%,同样具有良好的抗流感作用。但研究发现,GZP中大部分成分难溶于水,不利于药物的溶出和吸收。因此本文采用制剂学手段,采用一定技术对GZP进行处理后,再采用挤出滚圆技术制备成微丸增加药物的比表面积[6];并通过单因素试验法结合Box-Behnken效应面法,以溶出度为评价指标,对微丸的处方进行优化,以期提高药物溶解度,增加药物的溶出。
1 仪器与试药
CML湿法制粒挤出滚圆一体机(caleva);ZRS-8G智能溶出试验仪(天津精拓仪器科技有限公司);ACQUITY I-CLASS & PAD超高效液相色谱仪(美国Waters公司);SK7210HP KUDOS超声波清洗仪(上海科导超声仪器有限公司);METTLER TOLEDO万分之一分析天平、XS205 DualRang型分析天平(梅特勒-托利多国际贸易有限公司)。
吐温80(天津伦斯生物技术有限公司);甲醇(色谱纯,Fisher公司);甲酸(质谱纯,DikmaPure);乙酸铵(分析纯,北京化工厂);高纯水(Milli-Q超纯水系统制备);0.22 μm 微孔滤膜(津腾公司);聚维酮 K30(PVP K30)、微晶纤维素101(MCC 101)(安徽山河药用辅料股份有限公司);淀粉、乳糖、交联聚维酮(PVP-P)、低取代羟丙纤维素(L-HPC)、交联羧甲基纤维素钠(CCMC-Na)(北京凤礼精求医药股份有限公司);羧甲基淀粉钠(CMS-Na,西安天正药用辅料有限公司);羟丙基-β-环糊精(HP-β-CD,一鸣精细化工有限公司);抗坏血酸、D-异抗坏血酸钠[石药集团维生药业(石家庄)有限公司];磷酸二氢钾、氢氧化钠(分析纯,天津市光复科技发展有限公司)。
绵马贯众有效部位(自制,绵马贯众素ABBA含量为58.42%,批号:20201116);绵马贯众素ABBA(自制,纯度为94.20%)。
2 方法与结果
2.1 色谱条件
色谱柱:Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm);流动相:含0.1%甲酸、10 mmol·L-1乙酸铵的甲醇(A)-含0.1%甲酸、10 mmol·L-1乙酸铵的水(B),梯度洗脱(0~3 min,80%~84%A;3~12 min,84%~89%A;12~13 min,89%~100%A;13~19 min,100%~80%A);进样量:1 μL;流速:0.3 mL·min-1;检测波长:298 nm;柱温:40℃;样品室温度:25℃。
2.2 GZP微丸单因素考察
2.2.1 制备工艺因素考察 微丸中最常用的成型促进剂为MCC[7-8],本研究中采用MCC 101作填充剂,0.5% PVP K30水溶液为黏合剂,以微丸收率、休止角、丸条性状为评价指标,考察挤出速度(40、50、60、70 r·min-1)、滚圆速度(1200、1500、1800 r·min-1)、滚圆时间(3、5、7 min)对微丸成型的影响。结果表明,各挤出速度下微丸休止角均小于40°,说明微丸流动性满足要求,结合收率和生产效率选定挤出速度为60 r·min-1;当滚圆速度为1500 r·min-1时,微丸收率略高于其他组,且微丸粒径相对均一,因此选定1500 r·min-1作为滚圆速度;当滚圆时间为5 min和7 min时,微丸的粒径均较为均匀,但随着滚圆时间的延长,产生较多细粉,因此选定滚圆时间为5 min。
2.2.2 黏合剂浓度考察 微丸成型预试验中发现采用MCC与乳糖混合制备微丸时,微丸成型性较好,因此设定药物与填充剂的比例为1∶3,填充剂中MCC的比例为66.67%,以收率、休止角、微丸外观为评价指标,考察PVP K30水溶液作黏合剂时[9],其浓度对微丸成型的影响。结果显示,当黏合剂浓度为0.5%和1%时,收率和休止角无显著差异,说明黏合剂浓度对微丸成型无影响,因此根据微丸圆整度选定黏合剂为0.5% PVP K30水溶液。
2.2.3 崩解剂的种类和用量筛选 在微丸制备过程加入适量崩解剂,可以有效提高微丸崩解速率,从而提高药物溶出速率[10-13]。固定崩解剂用量为5%,以微丸收率、休止角、微丸外观为评价指标,分别考察崩解剂CMS-Na、PVP-P、L-HPC、CCMC-Na对微丸成型的影响。结果显示,各组微丸收率均高于90%,休止角均小于30°,流动性良好,说明崩解剂的种类对于微丸成型无影响,结合药物性质选用CMS-Na为崩解剂。分别制备CMS-Na用量为1%、5%和8%的微丸,置于37℃、100 r·min-1和900 mL含5%吐温80的水溶液中,以微丸崩解时间为指标,考察崩解剂的用量。结果显示,崩解剂用量对微丸崩解时间有显著影响,当崩解剂用量为5%和8%时崩解时间差异不明显,因此选定崩解剂用量为5%。
2.2.4 填充剂中MCC的比例考察 MCC作为骨架材料显著影响微丸后期释药速率,且乳糖作为致孔剂,能够显著提高释药速率[14]。因此选用MCC与乳糖为填充剂,以45 min时微丸的溶出度为评价指标,考察填充剂中MCC的比例为66.67%、50.00%、33.33%、20.00%时对微丸溶出度的影响。结果显示,随着MCC比例的降低,溶出度逐渐提高,但当MCC比例继续降低时微丸成型较差,因此初步确定填充剂中MCC的比例可选择20%左右。
2.2.5 增溶方法考察 参考文献[15-18]方法,考察HP-β-CD共研以及PEG 6000制备固体分散体对GZP的增溶效果。具体操作如下:
将HP-β-CD共混物(按质量比0.5∶1称取HP-β-CD与药物适量,混合均匀后于研钵中共研30 min)和固体分散体(称取3.00 g PEG 6000于80℃下水浴至熔融状态,逐量加入3.00 g GZP,搅拌均匀,迅速于-18℃下放置4 h,真空干燥过夜后,粉碎过80目筛)。按处方比例加入其他辅料,混合均匀后制备微丸,以45 min时微丸溶出度为评价指标,考察增溶方法。结果显示,HP-β-CD处理后的GZP微丸的溶出度超过50%,显著高于固体分散体组(27.14%),因此选用HPβ-CD共研法对药物进行预处理后,再制备微丸。
2.2.6 HP-β-CD与药物的比例考察 参考文献[19-20]对HP-β-CD与药物的比例进行考察。称取适量HP-β-CD与药物混合均匀并共研30 min预处理后,按表1比例加入其他辅料制备微丸,其中1-1组和1-2组微丸均可成型,而1-3组微丸无法成型。

表1 微丸处方参数及微丸性状评价Tab 1 Pellet formulation parameters and character evaluation
分别称取1-1组和1-2组微丸,以45、90、180 min时累积溶出度为评价指标,选定较为适宜的微丸处方。结果表明,两组微丸溶出行为较为接近,且45 min时1-1组微丸溶出度超过50%,因此结合微丸性状选定1-1组为较优处方。
2.3 微丸溶出度的测定
2.3.1 溶出介质的选择 根据前期试验结果,为保证达到漏槽条件,选择含5%吐温80的PBS溶液(pH=6.8)作为溶出介质,考察1-1组微丸的溶出情况,结果发现微丸的溶出度在后期呈现明显的降低趋势。由于GZP中含有较多的间苯三酚类成分,推测可能是由于其酚羟基发生氧化反应从而降解,因此考虑在溶出介质中加入适量抗氧剂[21]。
以药物的稳定效果为评价指标,对抗氧剂的种类和用量进行筛选,主要考察煮沸和加入不同量的抗坏血酸、D-异抗坏血酸钠的抗氧化效果。以溶出介质为溶剂,配制一定浓度的药物溶液,并加入抗坏血酸(5 mg·mL-1)、D-异抗坏血酸钠(5、2、1 mg·mL-1),于37℃,100 r·min-1条件下恒温水浴振摇,分别于0 min、15 min、30 min、45 min、1 h、2 h、3 h、4 h、6 h、8 h、10 h取样,续滤液进样,计算药物浓度。以时间为横坐标,测得浓度和0 min时药物浓度的比值为纵坐标绘制药物稳定性曲线图,结果见图1。2 mg·mL-1和5 mg·mL-1的D-异抗坏血酸钠对于药物的稳定效果较好,其中5 mg·mL-1组药物浓度相对更为平稳,因此选定抗氧化剂为5 mg·mL-1的D-异抗坏血酸钠。

图1 不同抗氧剂下的药物稳定性曲线图Fig 1 Stability curves of drugs with different antioxidants
2.3.2 溶出度的方法学研究
① 专属性考察:分别用溶出介质溶解对照品、样品和空白辅料,按“2.1”项下色谱条件,进样1 μL,记录色谱图,结果如图2所示,空白溶出介质和辅料对绵马贯众素ABBA的测定没有干扰。
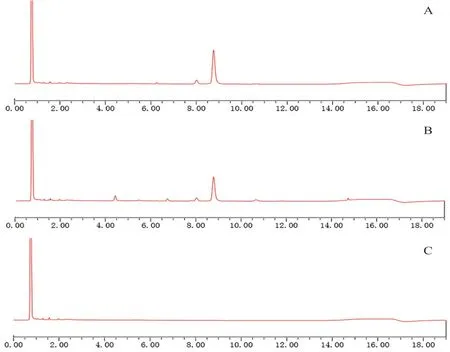
图2 绵马贯众有效部位微丸UPLC色谱图Fig 2 UPLC chromatogram of pellets from effective parts of Dryopteris crassirhizoma Nakai
② 精密度考察:取绵马贯众素ABBA对照品适量,溶出介质溶解,连续进样6次,RSD小于3%,说明精密度良好。
③ 线性考察:精密称定绵马贯众素ABBA 6.36 mg于25 mL量瓶中,加入溶出介质超声至溶解,定容,逐级稀释配制成质量浓度分别为2.41、4.82、9.63、48.17、96.34、143.79、191.72、239.64 μg·mL-1的系列溶液,按“2.1”项下色谱条件进样,记录色谱图,以峰面积对质量浓度进行线性回归,得到线性回归方程为y=8.112×103x-1.707×104(R2=0.9996)。表明绵马贯众素ABBA在2.41~239.64 μg·mL-1内与峰面积线性良好。
④ 样品稳定性考察:称取适量样品,溶出介质溶解,分别于0、2、4、6、8、10、12 h进样,保留时间和峰面积的RSD小于3%,说明样品在12 h内稳定性良好。
⑤ 回收率考察:精密称取样品约50%、80%、100%各3份,按处方比例加入空白辅料,溶出介质溶解,进样测定,计算回收率,低、中、高组的回收率平均值分别为97.25%、97.36%、95.90%,RSD均小于3%。
2.3.3 溶出度测定方法 根据2020年版《中国药典》四部通则0931溶出度测定第二法(桨法),以900 mL含5%吐温80和5 mg·mL-1D-异抗坏血酸钠的PBS溶液(pH=6.8)为溶出介质,溶出温度为(37±0.5)℃,转速为100 r·min-1,微丸投样量为0.3 g,分别于5 min、30 min时取样,并及时补加同温等体积溶出介质,0.22 μm微孔滤膜过滤,续滤液按“2.1”项下色谱条件进样,采用外标法以峰面积计算微丸累积溶出度。
2.4 Box-Behnken效应面法(BBD)优化微丸处方
2.4.1 因素水平表及试验结果 根据单因素考察结果,分析GZP微丸制剂工艺的影响因素和各因素所取水平,固定药物与填充剂的比例为1∶3,黏合剂为0.5% PVP K30水溶液,挤出速度60 r·min-1,滚圆速度1500 r·min-1,滚圆时间5 min,选取HP-β-CD与药物的比例(A)、填充剂中MCC的比例(B)、CMS-Na用量(C)作为考察因素,结合BBD设计原理,每个因素设定3个水平,见表2,按BBD试验设计表进行试验,以5 min时累积溶出度(Y1)和30 min时累积溶出度(Y2)作为评价指标,试验设计表与结果见表3。

表2 因素水平Tab 2 Factor and level
2.4.2 模型拟合 应用Design-Expert 8.0.5软件对表3中的数据进行处理,对各水平进行多元非线性回归,得到二项式拟合的方程分别为:Y1=6.10+10.39A-5.18B+0.17C-7.55AB+0.51AC-0.46BC+10.76A2+8.82B2-7.19C2;Y2=19.95+18.64A-6.26B+1.05C-9.24AB+1.52AC-1.24BC+14.76A2+11.96B2-8.57C2;Y1和Y2的回归方程的P值均小于0.05,说明其多元回归拟合模型成立。
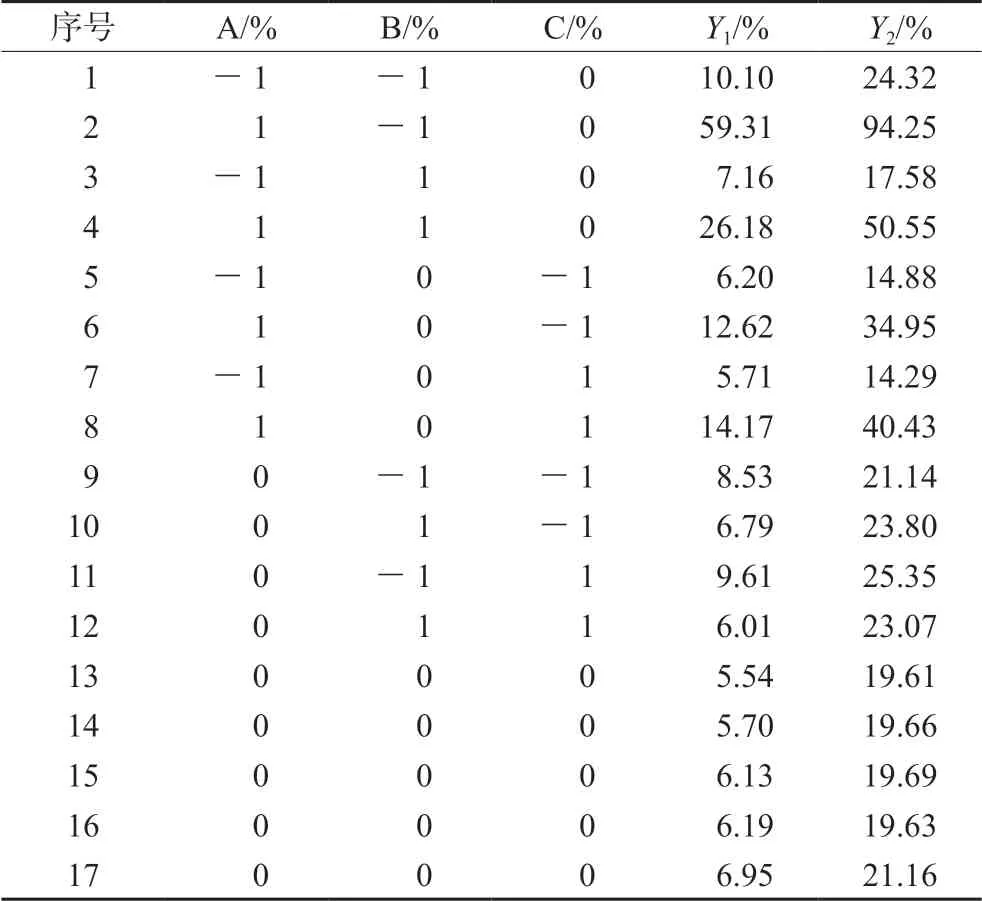
表3 BBD试验设计与结果(n=2)Tab 3 BBD test design and results (n=2)
2.4.3 效应面优化与预测 应用Design-Expert 8.0.5软件分别绘制Y1、Y2对两两因素的三维效应面图,直观描绘出各因素的影响,分别见图3。鉴于本研究的目的是提高药物的溶出度,因此在评价指标中,选择释放速度越快越好。故设定Y1(5 min时累积溶出度)的取值范围为50%~80%,以反映微丸的快速释放过程;Y2(30 min时累积溶出度)的取值范围为80%~100%,以反映微丸是否能够释放完全,将各效应面对应的等高线进行叠加绘制二维等高线叠加图,见图4,图中黄色区域为理想的较优区域,此时CMS-Na用量(C)固定为5%。
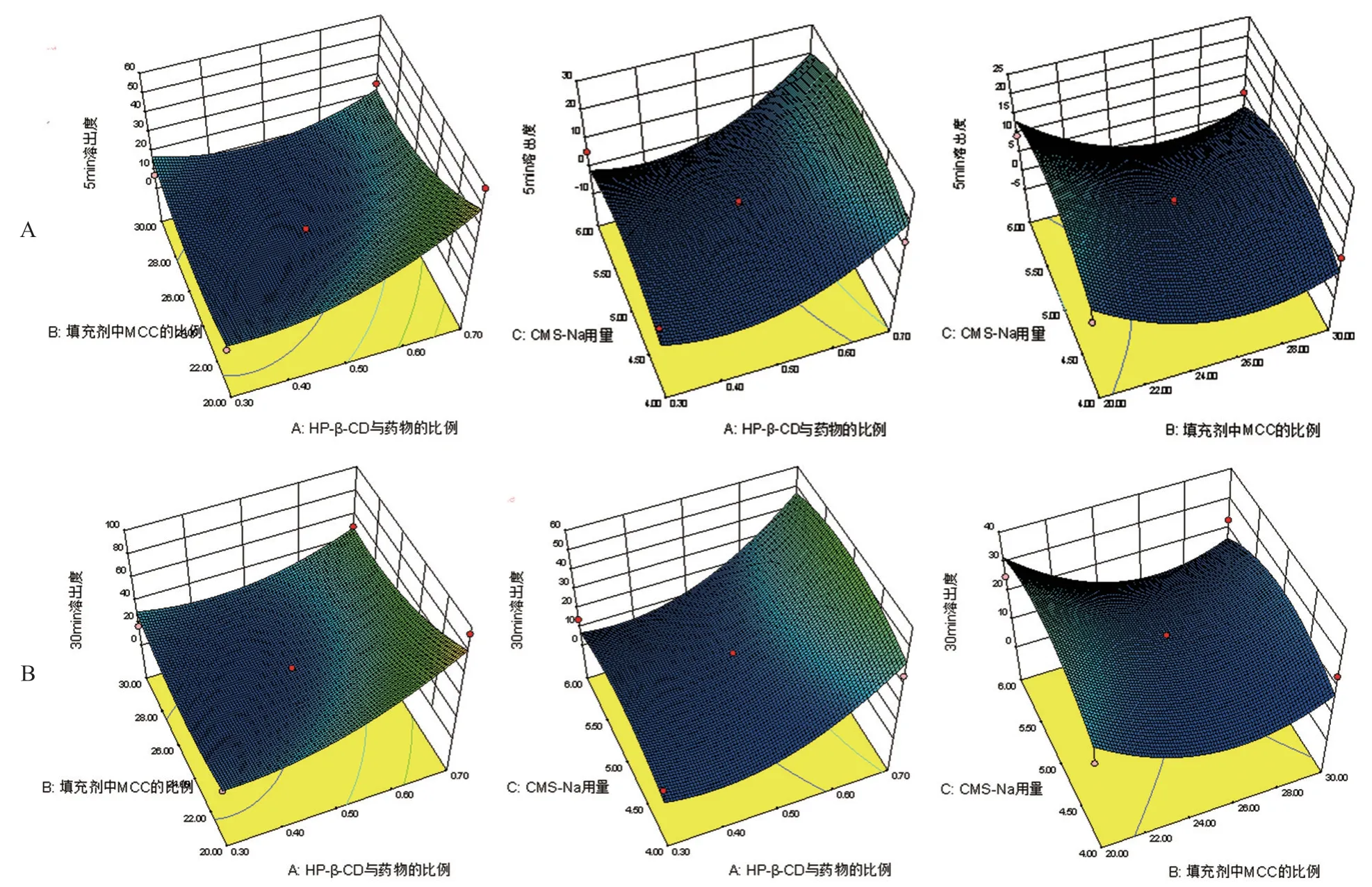
图3 各因素对Y1(A)及Y2(B)影响的三维效应面图Fig 3 Effect of each factor on Y1(A)and Y2(B)three-dimensional effect surface diagram
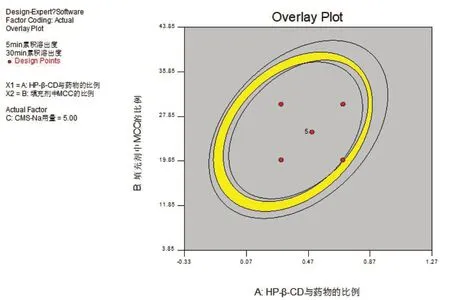
图4 处方优化的二维等高线叠加图Fig 4 Two-dimensional contour stacking diagram of prescription optimization
2.5 处方验证
根据试验优化结果,结合实际情况,选择上述优化区域中的处方,各因素为:HP-β-CD与药物比例(A)为0.70,填充剂中MCC比例(B)为18.50%,CMS-Na用量(C)为5%,进行微丸的制备,并分别测定微丸在5、30 min时累积溶出度,平行测定6组。结果如表4所示,Y1和Y2均满足试验预设取值范围,且RSD值均小于1%,说明该处方稳定合理。
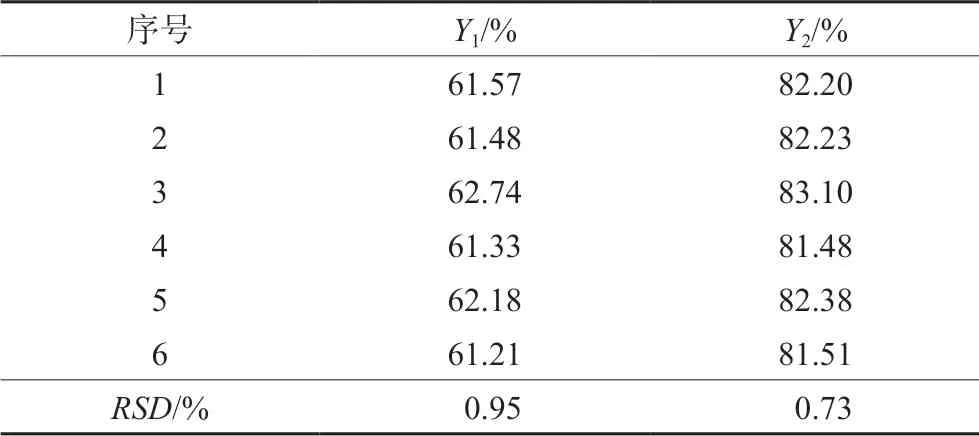
表4 处方验证的溶出度测定结果Tab 4 Dissolution test results of prescription verification
3 讨论
绵马贯众有效部位主要为水难溶性成分,现代制剂研究中对于难溶性成分主要采用滴丸、微丸等制剂形式通过增加药物的比表面积以改善其溶解度和溶出速度,其中微丸的工艺技术较为成熟,因此本文采用挤出滚圆技术制备微丸。
MCC是制备微丸的常用辅料,是微丸成型促进剂,但MCC易形成骨架材料,乳糖与之搭配使用,可以在骨架中迅速溶解形成孔道,吸引水分进入微丸内部,并加入适量崩解剂进一步加快微丸崩解速度,从而促进药物的释放。鉴于药物难溶于水的性质,采用羟丙基-β-环糊精对药物预处理后,使得微丸溶出度得到显著提升。
溶出度研究中发现药物成分在溶液中稳定性较差,因此在溶出介质中加入适量抗氧剂,使其稳定性增强。并在此基础上采用二维等高线叠加及Box-Behnken效应面法选定最优处方工艺区域,对于处方的选择具有良好的指导作用。
在后续的研究中,将对羟丙基-β-环糊精共研产物进行更为深入的研究,从而观察药物的具体存在状态,探索羟丙基-β-环糊精共研法增溶原理。
