镁合金表面PEF 膜与PEO 膜腐蚀防护行为的对比研究
2021-07-03齐玉明刘思勤罗兰彭振军梁军王鹏
齐玉明,刘思勤,罗兰,彭振军,梁军,王鹏
(1.中国科学院兰州化学物理研究所 固体润滑国家重点实验室,兰州 730000;2.中国科学院大学 材料与光电研究中心,北京 100049)
镁合金是密度最小的金属结构材料,具有良好的力学性能、工艺性能以及生物学性能,被广泛应用于国防工业、航空航天、汽车工业、3C 产品、生物医用材料等诸多领域。当下,伴随着新能源汽车行业发展的浪潮,以铝、镁合金材料为代表的轻质材料的研究与应用,较传统汽车行业更为迫切和重要[1-2]。然而由于较高的化学活性,镁合金极易发生腐蚀,这也是推广镁合金大规模应用的主要限制之一。
对于镁合金的腐蚀防护,等离子体电解氧化(PEO)是有效的表面处理技术之一。该技术一般都采用弱碱性的水溶液作为电解液,通过等离子体电解反应,在镁合金表面形成氧化物陶瓷膜层。PEO 膜可以作为屏障,有效抵御腐蚀介质对镁合金基体材料的腐蚀破坏,同时兼具较高的硬度、良好的耐磨性和优异的涂挂性[3-4]。此外,镁合金PEO 膜主要由氧化物陶瓷相组成,保持甚至提升了镁合金基体材料的生物学性能[5-6]。
尽管镁合金PEO 膜可以有效地改善基材的耐蚀性,但是在长效耐蚀性和耐酸腐蚀等方面的表现仍然有待提高。研究表明[7],在中性腐蚀介质中,镁合金PEO 膜容易因MgO 的水合反应而失效,不足以提供长效的保护作用。而在酸性腐蚀介质中,酸的中和作用将极大地加速膜层失效[8]。研究表明,由于MgF2的化学稳定性高于MgO,通过在膜层中引入氟化物可以有效地改善膜层的耐蚀性[9-10]。目前,在PEO 膜中引入氟化物的主要途径有:一是,在以含氧酸盐为主盐的电解液中加入含氟化合物(如KF、K2ZrF6等)添加剂[9-10];二是,直接将含氟化合物(如KF、K2ZrF6、K2TiF6等)作为电解液主盐[11-13]。然而,由于电解液中水的作用,这些PEO 膜的组成仍然以基材的氧化物为主。
本课题组开发了NH4F-EG 室温非水电解液,并在镁合金表面实现了等离子体电解氟化(PEF)过程[14-15]。初步研究表明,PEF 膜主要由基材的氟化物构成,具有不同于PEO 膜的多孔结构,在中性和酸性腐蚀环境中都对镁合金基材具有良好的保护作用。
本文通过电化学测试、浸泡实验和盐雾实验,系统评价了镁合金表面新型PEF 膜的防腐蚀性能,对比研究了PEF 膜与PEO 膜在腐蚀防护行为上的异同,同时基于现有镁合金PEO 膜的腐蚀防护机理,揭示了PEF 膜的腐蚀防护失效机制。
1 实验
1.1 膜层的制备
基体材料为AZ91D 镁合金,其化学组成(以质量分数计)为:Al 8.5%~9.5%,Zn 0.45%~0.95%,Mn 0.17%~0.4%,Si ≤0.08%,Fe ≤0.004%,Cu ≤0.02%,Ni ≤0.001%,余量为Mg。镁合金试样的规格为25 mm×25 mm×8 mm,经SiC 水砂纸逐级打磨,抛光至2000 目,随后分别经去离子水和丙酮超声清洗,再经去离子水冲洗后,吹干备用。
PEF 电解液为NH4F 的乙二醇溶液,NH4F 的质量浓度为80 g/L。NH4F(烟台双双化工有限公司)和乙二醇(EG,利安隆博华(天津)医药化学有限公司)均为分析纯。在25 ℃下,该电解液的电导率和pH 值分别为5.35 μS/cm 和7.42。
PEO 电解液为Na3PO4·12H2O(分析纯),和NaOH的水溶液,二者质量浓度分别为19、1 g/L,分别由成都市科隆化学品有限公司和利安隆博华(天津)医药化学有限公司提供。在25 ℃下,该电解液的电导率和pH 值分别为16.72 μS/cm 和14.37。
等离子体电解过程采用双极性脉冲微弧氧化电源(JHMAO200H,北京金弧绿保科技开发有限公司),其额定功率为200 KW。该过程中,试样连接电源正极作为阳极,不锈钢管连接电源负极作为阴极,管内通自来水起到冷却电解液的作用,通过外接磁力搅拌器的机械搅拌作用保证电解液浓度和温度的均匀性。PEF 和PEO 均采用相同的电参数:频率、占空比和正负脉冲比分别为100 Hz、20%和1:5,正负电流密度分别设定为8、0 A/dm2,电解液温度维持在(25±2) ℃。
PEF 和PEO 处理过程的时长、相应过程的终止电压以及膜层厚度如表1 所示。其中,膜厚测量采用了涡流测厚仪(Elektro Physik Mini Test 1100),将10 个随机测试点的均值作为膜厚值。

表1 PEF 与PEO 过程处理时长、终止电压与膜层厚度Tab.1 Processing time, final voltage and resultant coating thickness of PEF and PEO processes
1.2 膜层的表征
采用扫描电子显微镜(SEM,JEOL JSM-5600LV)观察膜层的表面和截面形貌,并通过X 射线能量色散谱仪(EDS,Oxford Instruments X-MaxN 80)分析膜层的化学组成。通过X 射线衍射仪(XRD,PANalytical Empyrean),在掠入射模式下分析膜层的物相组成,阳极靶材选用Cu 靶(Kα1=0.154 06 nm),入射角固定为2°,扫描步长为0.1°,扫描范围为20~80°。
基于膜层表面形貌的SEM 照片,通过软件Image J(Ver. 1.52,NIH Image)对膜层表面的微孔进行统计分析。
采用电化学工作站(M e t r o h m A u t o l a b PGSTAT302N)测试了膜层的电化学腐蚀行为。测试采用三电极体系,试样、饱和Ag/AgCl 和铂片分别为工作电极、参比电极和辅助电极。中性腐蚀介质为5%的氯化钠(NaCl,天津市百世化工有限公司)水溶液;酸性腐蚀介质是以中性腐蚀介质为基础,通过冰醋酸(CH3COOH,天津北辰方正化学试剂厂)酸化至pH=3 的酸性溶液,同时符合QB/T 3827—1999《轻工产品金属镀层和化学处理层的耐腐蚀试验方法 乙酸盐雾试验(ASS)法》。电化学测试均在室温下进行,试样暴露面积为1 cm2。在试样暴露区域与腐蚀介质反应30 min 后,进行动态电位极化曲线测试。所有测试均重复三次,以保证实验结果的重复性和可信度。
采用浸泡实验和盐雾实验评估了膜层的长效腐蚀防护性能和宏观腐蚀行为。预先用可剥离树脂封装试样的棱角和侧面(即25 mm×8 mm 面),仅暴露测试表面(即25 mm×25 mm 面)的中间区域,其面积约为20 mm×20 mm。浸泡实验设置2 个平行试样,其4 个暴露面均为测试面,腐蚀介质与电化学测试实验相同。酸性盐雾实验所采用的酸液与电化学测试所用的酸液相同,具体参照标准QB/T 3827—1999《轻工产品金属镀层和化学处理层的耐腐蚀试验方法 乙酸盐雾试验(ASS)法》进行。
通过接触角测量仪(KRUSS DSA100)测试膜层对水的接触角时,随机选取5 个测试点进行测量,求其均值作为接触角数值。
2 结果及分析
2.1 膜层的组成表征
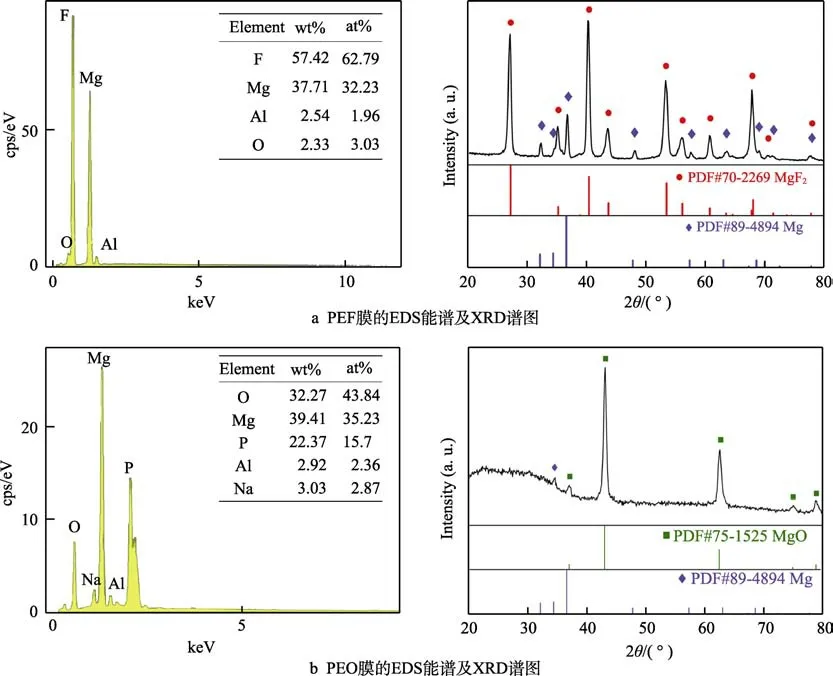
图1 PEF 膜与PEO 膜的化学组成及物相分析Fig.1 Chemical composition and phase analysis of PEF and PEO: a) EDS spectrum and XRD spectrum of PEF; b) EDS spectrum and XRD spectrum of PEO
两组膜层的元素含量和物相组成如图1 所示。图1a中的EDS 测试结果表明,PEF 膜主要由F 和Mg 组成,并含有极少量的Al 和O,但是NH4F-EG 电解液中的C 和N 并没有在膜层中检出。其中,F 与Mg 的含量(以质量分数计)分别高达57.42%和37.71%,而Al 和O 均低于3%。通过计算可以发现,该膜层中的F/Mg 比率为1.95,与MgF2的组成极为接近。因此可以推断出,该膜层的主要物相组成是MgF2。从PEF 膜的XRD 图谱可以看出,衍射信号主要来自于MgF2,表明膜层中主要物相为MgF2,由于X 射线穿透膜层,检测到了来自于基材金属中的Mg 信号。这也与EDS 的分析结果一致,表明膜层中的F 和Mg结合形成了化合物MgF2。由于Al 和O 的含量都很低,相应化合物的衍射信号并没有在XRD 图谱中出现。
由图1b 可知,镁合金PEO 膜中的元素组成以Mg、O 和P 为主,并含有少量的Al 和Na。其中Mg为39.41%,O 和P 分别高达32.27%和22.37%,而Al 和Na 均在3%左右。XRD 图谱表明,衍射信号主要来自于MgO,同时在20°~37°范围内,出现了明显的漫反射信号(鼓包)。通常在磷酸盐电解液中的P会以非晶相的形式存在于镁合金PEO 膜中[16-17]。结合元素分析结果可知,此处的漫反射信号表明,P 形成的非晶相化合物存在于膜层中。由此可见,镁合金PEO 膜的主要物相组成是MgO,并存在一些含磷的非晶相化合物。
2.2 膜层的微观结构表征
两组膜层的表面形貌如图2 所示,可见二者都具有多孔结构。从图2a 可以看出,PEF 膜表面的微孔较为细长,将膜层表面分隔为大量的数微米的区域,同时PEF 膜表面最大微孔直径不超过5 μm,并且随机分布着一些数微米甚至纳米级的微孔和微凸起。从图2b 中可以看出,PEO 膜表面的孔隙更趋近于椭圆,其中较大的孔隙不足以将膜层分隔成为相对独立的微区,较小的孔隙则随机分布在膜层表面。同时,PEO膜表面最大微孔直径高达40 μm,且随机分布有一些数微米的微孔。

图2 膜层表面的微观结构Fig.2 Surface morphologies of coatings: a) PEF coating; b) PEO coating
图3 为PEF 和PEO 膜表面微孔的统计分析结果。PEF 膜的表面孔隙率为15.11%,其单位面积上的孔隙数量高达5.311×107/cm2;而PEO 膜的表面孔隙率仅为 6.32%,其单位面积上的孔隙数目也仅为2.44×106/cm2。PEF 膜的表面孔隙率高达传统PEO 膜的2.5 倍,单位面积上的孔隙数量则高出了一个数量级。统计发现,PEF 膜表面最大孔隙的面积仅为平方微米级,而PEO 膜的则为数百平方微米级;这也与图2 中观察到的结果一致。对于PEF 膜而言,极大(μm2级)和极小(10–4μm2级)的孔隙所占比例分别为6.13%和8.81%,10–1、10–2、10–3μm2级孔隙的占比分别为34.10%、31.03%和19.92%。对于PEO 膜而言,尺度越小的孔隙数量越多,极大(102μm2级)和极小(10–2μm2级)的孔所占比例分别为0.42%和59.91%,101、100、10–1μm2级孔隙的占比分别为2.49%、11.47%和26.43%。

图3 膜层表面孔隙的统计分析Fig.3 Statistical analysis of the pores on the surface of the coating: a) surface porosity; b) number of micro-pores; c) area of micro-pores on PEF coating; d) area of micro-pores on PEO coating
两组膜层的截面形貌及相应的元素分布如图4所示。从图4a 中可以看出,PEF 膜内部也是多孔结构,孔隙尺度不超过5 μm,数量较多,形状不规则,在膜层厚度方向上的各处均有分布。相应的元素分布图表明,膜层内部的组成主要是Mg 和F,元素分布均匀,没有分层现象。从图4b 中可以看出,PEO膜在结构上具有明显的分层现象。外层为多孔层,孔隙尺度较大(~10 μm),形状较为规则;在膜层与基材的界面附近为致密层,其厚度远小于外部多孔层。相应的元素分布图表明,膜层内部的主要组成元素是Mg、O 和P,元素分布均匀,没有分层现象。对比两组膜层的截面图,可见二者的膜厚相当,均在10 μm 左右,与表1 中通过测厚仪测得的数据一致。

图4 膜层截面微观结构Fig.4 Cross-sectional morphologies of coatings: a) PEF coating; b) PEO coating
2.3 膜层在中性环境中的腐蚀行为
AZ91 镁合金基材及两组膜层在中性腐蚀介质中的电化学腐蚀行为结果如图5 所示。开路电位随时间的变化曲线如图5a 所示。可见,在起初的0.25 h 内,AZ91 镁合金基材的开路电位正移至–1530 mV 左右,并在随后的0.5 h 内逐渐负移至–1560 mV 左右,而后进入稳定状态,开路电位在(–1570±20) mV 内小幅波动。在起初的0.25 h 内,PEO 膜的开路电位正移至–1510 mV 左右,随后开始负移,并于1.17 h 时达到–1630 mV 左右,随后又逐渐正移,并于4.7 h 时从–1460 mV 左右骤然负移至–1616 mV 左右。随后在AZ91 基材的曲线两侧波动,且幅度逐渐减小,最终与之重合,表明膜层失效。在起初的6.2 h 内,PEF膜的开路电位逐渐正移至–1460 mV 左右,随后快速负移至与AZ91 基材的曲线重合,表明膜层失效。
图5b 为动态电位极化测试的结果,其相应的塔菲尔(Tafel)线性拟合结果列于表2。可见PEO 膜的自腐蚀电位(Ecorr)较AZ91 镁合金基材正移了69 mV,自腐蚀电流密度(Jcorr)较AZ91 镁合金基材降低了2个数量级。PEF 膜的自腐蚀电位(Ecorr)较AZ91 镁合金基材正移了124 mV,较PEO 膜正移了55 mV;自腐蚀电流密度(Jcorr)较AZ91 镁合金基材降低了2个数量级,较PEO 膜层提高了1 倍。
通过浸泡实验,研究了两组膜层的宏观腐蚀行为,结果如图6 所示。发现二者的腐蚀形式均为局部腐蚀。当浸泡时间达到168 h 时,PEF 膜表面尚未发生明显变化,而PEO 膜上已经出现了腐蚀坑(如圆圈所示)和白色鼓包(如箭头所示)。当浸泡时间达到504 h 时,PEF 膜表面出现了腐蚀坑(如圆圈所示);而PEO 膜上的局部区域已经发生了严重的腐蚀破坏,在腐蚀坑周围形成并堆积有白色的腐蚀产物,白色鼓包的数量增多,尺寸也有所增大。随着浸泡时间的进一步延长,腐蚀坑的面积增大,腐蚀产物增多,其他区域也逐渐发生了轻微腐蚀的痕迹。相较而言,PEF膜表面腐蚀痕迹的发展速率更慢。浸泡1128 h 后,可以看到两组膜层表面的腐蚀产物均覆盖了腐蚀坑,遏制了其在膜层表面的进一步扩展。结果表明,相较于传统PEO 膜,PEF 膜在中性腐蚀介质中具有更加优异的长效耐蚀性能。

表2 中性腐蚀介质中动态电位极化测试的拟合结果Tab.2 The calculated result of the potentiodynamic polarization test in neutral corrosive medium
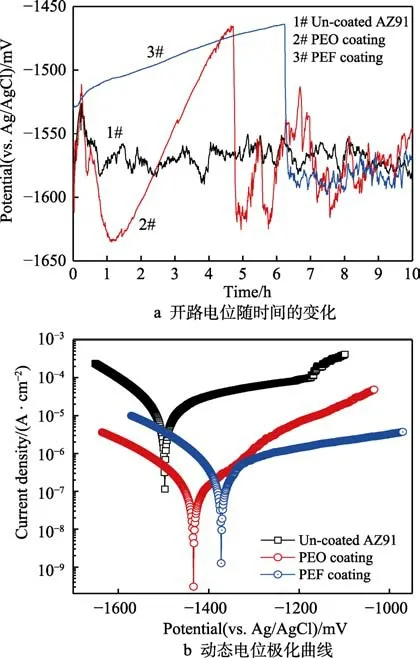
图5 中性腐蚀介质中的电化学实验Fig.5 Electrochemical tests in neutral corrosive medium: a)open circuit potential with immersion time; b) potentiodynamic polarization test

图6 中性腐蚀介质中的浸泡实验Fig.6 Immersion test in neutral corrosive medium
中性浸泡实验结束(1128 h)后,PEF 膜和PEO膜的腐蚀形貌如图7 所示。对于PEF 膜而言,宏观上尚未出现腐蚀痕迹的区域的微观形貌如图7a 所示。可见其在结构上尚未发生明显的变化,在组成上,F 含量有所减小,而O 含量小幅增大。腐蚀坑附近的微观形貌如图7b 所示,其中A 区为尚未出现腐蚀的区域,B 区为宏观上的“白色鼓包”(即堆积的腐蚀产物),C 区为剥去腐蚀产物后的区域。通过相应的元素分布图可以看到,C 区已经发生了严重的腐蚀,腐蚀介质穿透并破坏了膜层,在金属基材表层形成了较深的腐蚀坑。B 区和C 区的元素分布没有显著差异,表明腐蚀产物表层与内部的组成是一致的。
对于PEO 膜而言,宏观上尚未出现腐蚀痕迹的区域的微观形貌如图7c 所示,可见其在结构上尚未发生明显的变化,在组成上,P 含量有所减小,而O含量有所增大。腐蚀坑附近的微观形貌如图7d 所示,其中A 区所示为尚未出现腐蚀的区域,B 区所示为宏观上的“白色鼓包”(即堆积的腐蚀产物),C 区所示为剥去腐蚀产物后的区域。通过相应的元素分布图可以看到,C 区已经发生了严重的腐蚀,腐蚀介质穿透并破坏了膜层,在金属基材表层形成了较深的腐蚀坑。对比B 区和C 区的元素分布可以看到,表层的腐蚀产物中含有较多的Cl。对比A 区和B、C 区的元素分布可以看到,腐蚀产物中的O 含量显著高于原始PEO 膜,但是其中几乎不含有P。

图7 PEF 膜和PEO 膜在中性腐蚀介质中浸泡1128 h 后的腐蚀形貌及元素分布Fig.7 Morphologies and element distribution of PEF and PEO coatings after immersion in neutral corrosive medium for 1128 h:a) no corrosion marks are found on the surface of the PEF coating; b) corrosion pits on the surface of PEF film and its surrounding area; c) no corrosion marks are found on the surface of the PEO coating; d) corrosion pits on the surface of PEO film and its surrounding area
2.4 膜层在酸性环境中的腐蚀行为
AZ91 镁合金基材及两组膜层在酸性腐蚀介质中的电化学腐蚀性能测试结果如图8 所示。开路电位随时间的变化曲线如图8a 所示,可以看到,AZ91 镁合金基材的开路电位始终稳定在–1600 mV 左右。在起初的1.6 h 内,PEO 膜的开路电位负移至–1800 mV 左右,随后在4.7 h 时,正移至与基材的曲线相重合,表明膜层完全失效。在起初的1 h 内,PEF 膜的开路电位正移至–1530 mV 左右,随后在1.5 h 时,负移至与基材曲线相重合,表明膜层完全失效。
图8b 为动态电位极化测试的结果,相应的Tafel线性拟合结果列于表3。可见,PEO 膜的自腐蚀电位较AZ91 镁合金基材正移了18 mV,自腐蚀电流密度较AZ91 镁合金基材降低了3 个数量级。PEF 膜的自腐蚀电位较AZ91 镁合金基材正移了71 mV,较PEO膜正移了53 mV;自腐蚀电流密度较AZ91 镁合金基材降低了2 个数量级,较PEO 膜层高了1 个数量级。
市场化程度(mart):樊纲等则在问卷数据的基础上,通过构建市场化评价指标体系,计算出市场化指数[8]4-7。鉴于数据的可得性,本文采用非国有固定资产投资占全社会固定资产投资的比重来衡量市场化水平。

图8 酸性腐蚀介质中的电化学实验Fig.8 Electrochemical tests in acidic corrosive medium: a) open circuit potential with immersion time; b) potentiodynamic polarization test
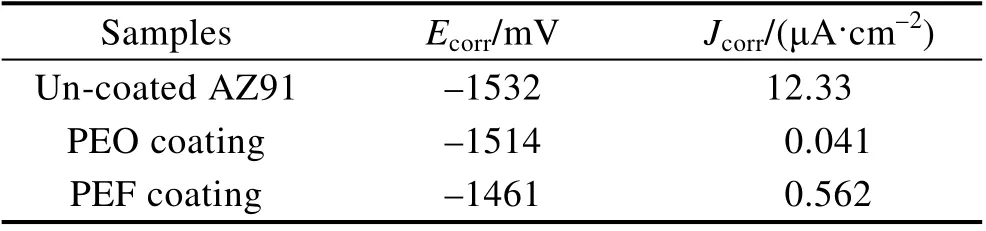
表3 酸性腐蚀介质中动态电位极化测试的拟合结果Tab.3 The calculated result of the potentiodynamic polarization test in acidic corrosive medium
通过浸泡实验研究了两组膜层的宏观腐蚀行为,结果如图9 所示。可以看出,在24 h 时,二者都出现了肉眼可见的腐蚀坑,整个过程中都没有观察到腐蚀产物的堆积。PEF 膜表面的腐蚀坑数量较少、面积较大;PEO 膜表面的腐蚀坑数量较多、面积较小。实际上,在3 h 时,PEO 膜表面就已经出现了一些细微的深色纹理,随着浸泡时间延长,该现象在14 h 时尤为明显。然而在腐蚀坑出现之前,PEF 膜的宏观形貌并未出现明显的变化。

图9 酸性腐蚀介质中的浸泡实验Fig.9 Immersion test in acidic corrosive medium
酸性浸泡实验中,试样的质量损失情况如图10所示。可见,经PEF 和PEO 处理的两组试样在浸泡过程中均发生了质量损失,二者的质量损失量随浸泡时间延长而持续增大。在同一时间节点上,经PEO处理的试样的质量损失高于经PEF 处理的试样。试样的质量损失反映了其表面膜层或基材的溶解情况。因此,在酸性腐蚀介质中,经PEF 处理的试样较PEO处理的试样溶解更慢,耐蚀性更好。
酸性浸泡实验结束(28 h)后,PEF 膜和PEO 膜表面的腐蚀形貌如图11 所示。图11a 中显示了PEF膜表面尚未出现腐蚀坑的区域的微观形貌。可见,其在结构上没有发生显著变化,只是多了一些颗粒物;在组成上,O 和Al 的含量都显著增多。腐蚀坑附近的微观形貌如图11b 所示,可见坑内(右侧)有大量团聚颗粒状结构,元素分布图表明其主要组成为Mg、O 和Cl,几乎没有F。表明该区域已经发生了严重的腐蚀,腐蚀介质穿透并破坏了膜层,在金属基材表层形成了较深的腐蚀坑。
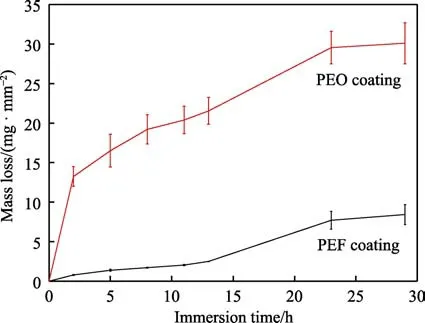
图10 酸性浸泡实验中样品的质量损失Fig.10 Mass loss of the PEF and PEO coated samples during the immersion test in acidic corrosive medium
图11c 反映了PEO 膜表面尚未出现腐蚀坑的区域的微观形貌,可见膜层发生了大面积的龟裂和剥落,产生了大量的微裂纹,在组成上,Al 含量显著增多,而P 含量显著减少。腐蚀坑及其附近区域的微观形貌如图11d 所示,可见已经形成了深度较大的沟壑。相应的元素分布图显示,在腐蚀坑内几乎没有探测到任何元素,这表明腐蚀坑的深度较大,内部堆积的腐蚀产物较少。由此可见,腐蚀坑形成后的腐蚀产物也几乎完全溶解于腐蚀介质中,没有在膜层表面或是腐蚀坑内堆积。
酸性浸泡实验中,PEO 膜与PEF 膜表面出现腐蚀坑的时间点没有显示出显著的差异。作为补充,通过酸性盐雾实验进一步考察了二者在酸性腐蚀介质中的宏观腐蚀行为,实验结果如图12 所示。可以看出,在96 h 时,PEF 膜表面出现白色突起,312 h 时,白色突起的数量增多并且出现了细小的腐蚀点。而PEO 膜表面在24 h 时就出现了细小的腐蚀点和白色突起,随着暴露时间延长,腐蚀点和白色突起的数量增多,一些腐蚀点扩展,形成了尺寸较大的腐蚀坑;当暴露时间达到312 h 时,PEO 膜被完全腐蚀破坏。
酸性盐雾实验312 h 后,PEF 和PEO 膜表面的腐蚀形貌如图13 所示。图13a 中显示了PEF 膜表面尚未出现明显腐蚀痕迹的区域的微观形貌,可见其表面有一些数十微米大小的颗粒物。相应的元素分布图表明,这些颗粒物中含有大量的O,而其中的F 含量则远低于邻近微区。通过对图13a 所示的这一类区域进行元素含量分析发现,其中的F 含量显著降低,而O含量显著升高。这说明此类区域也在一定程度上受到了酸性腐蚀介质的影响。图13b 中显示了PEF 膜上腐蚀坑及其附近区域的微观形貌。可见,腐蚀坑内(上方)出现了块状和颗粒状的腐蚀产物,其组成中含有大量的O。腐蚀坑外(下方)的膜层尚未发生腐蚀破坏,其组成和结构均未发生显著变化。
图13c 所示为PEO 膜表面尚未出现明显腐蚀痕迹的区域的微观形貌,可见膜层发生了大面积的龟裂,出现了大量的微裂纹,并局部脱落。同时,在其表面几乎看不到原始PEO 膜层的多孔结构。元素分析表明,膜层中的Al 含量显著增多,P 含量显著较少。这表明,在酸性腐蚀介质的作用下,膜层表面原有的孔隙被封堵。图13d 所示为PEO 膜表面腐蚀坑附近区域的微观形貌。可见,相较于腐蚀坑外(右侧),坑内(左侧)的O 含量更高,而P 含量更低。

图11 PEF 膜和PEO 膜在酸性腐蚀介质中浸泡28 h 后的腐蚀形貌Fig.11 Morphologies and element distribution of PEF and PEO coatings after immersion in acidic corrosive medium for 28 h: a)no corrosion marks are found on the surface of the PEF coating; b) corrosion pits on the surface of PEF film and its surrounding area; c) no corrosion marks are found on the surface of the PEO coating; d) corrosion pits on the surface of PEO film and its surrounding area

图12 酸性盐雾实验Fig.12 Acidic salt spray corrosion test

图13 PEF 膜和PEO 膜在酸性盐雾实验312 h 后的腐蚀形貌Fig.13 Morphologies and element distribution of PEF and PEO coatings after 312 h acidic salt spray corrosion test: a) no corrosion marks are found on the surface of the PEF coating; b) corrosion pits on the surface of PEF film and its surrounding area; c) no corrosion marks are found on the surface of the PEO coating; d) corrosion pits on the surface of PEO film and its surrounding area
3 讨论
目前,已经有大量的工作研究了镁合金PEO 膜的腐蚀防护行为及其失效机制,并形成了较为完备的理论体系。研究者普遍认为[18-21],PEO 膜层在金属基材表面形成了屏障,一方面在空间上隔绝了腐蚀介质与金属基材的接触,进而延缓了腐蚀的发生;另一方面,由于膜层本身的介电特性在很大程度上阻断了金属基材与外界环境的电荷交换,进而阻止了腐蚀的发生。相应地,膜层的腐蚀防护失效主要可归结为两种机制:一是膜层与腐蚀介质的相互作用,致使膜层本身被破坏而防护失效[8,22];二是腐蚀介质穿透膜层到达基材表面,与之发生相互作用,致使基材腐蚀[23-24]。但需要注意的是,这两类腐蚀防护失效机制往往同时存在于具体的腐蚀过程中,只是因膜层特性、腐蚀介质特性、测试方法等因素而有主次之分[25-27]。以上腐蚀理论不仅适用于PEO 膜,同时也适用于热喷涂涂层[28]、转化膜[29-30]和各种氧化膜[21]等的腐蚀过程,具有较为广泛的适用性。
基于以上理论可知,膜层完全失效的判据在于基材发生腐蚀,而基材发生腐蚀的一个必要条件在于腐蚀介质到达基材表面。对于电化学测试、浸泡实验和盐雾实验等腐蚀评价方法而言,腐蚀介质一方面通过微观孔隙向膜层深处扩散,另一方面与膜层物质发生化学反应。对于电化学测试中的动态电位极化测试而言,在系统的平衡电位附近进行电位扫描,相当于施加外场,迫使基材与腐蚀介质之间进行电荷交换,进而发生腐蚀反应。显然,此过程相当于加剧了腐蚀介质在膜层内部的扩散速率。相较于中性介质的渗透作用,酸性腐蚀介质对于膜层化合物的溶解和消耗,相当于大幅缩短了扩散路程[24],同样可以看作是加快了扩散速率。
腐蚀介质的扩散过程受膜层表面与内部孔隙结构分布情况的影响较大。其中,微裂纹和局部大尺度的微孔等缺陷都将构成腐蚀介质扩散的捷径,进而加速局部扩散和局部腐蚀的进程。膜层微观结构(图2和图4)以及微孔统计分析结果(图3)显示,PEF膜表面的微孔尺度小且分布均匀,但孔隙率较高,几乎没有微裂纹缺陷,内部孔隙之间的连通程度较低,连通孔的尺度较小。PEO 膜表面的微孔尺度较大,且随机分布有一些大尺度微孔,有微裂纹缺陷,内部孔隙之间的连通程度较高,连通孔的尺度较大。这也就表明,PEO 膜层中更易形成腐蚀介质扩散的捷径,进而加速了在局部区域内腐蚀坑的形成。
图14 为两组膜对水的润湿特性,可见水滴在PEF表面的接触角仅为14.2°,在PEO 膜表面的接触角为65.4°,表明PEF 膜的亲水性更好。由此可知,腐蚀介质在PEF 膜的表面更容易润湿,在其内部也将更易于扩散。然而各组宏观腐蚀实验结果均表明,PEF膜表面出现腐蚀痕迹的时间点均晚于PEO 膜。这就表明,尽管腐蚀介质在PEF 膜层内部的扩散速率较快,但在PEO 膜中扩散捷径处的扩散速率更快。

图14 水在膜层表面的接触角Fig.14 Water contact angles on PEF and PEO coatings
腐蚀介质与膜层之间的第一种相互作用是水合反应。通过对膜层腐蚀后尚未出现腐蚀痕迹的区域进行元素分析发现,经历中性腐蚀介质的作用后,PEF膜中的O 含量小幅增加,而在PEO 膜中则大幅增加;经历了酸性腐蚀介质的作用后,两组膜层中O 含量的增幅均较小。这表明两组膜层中的化合物均发生了一定程度的水合反应,其中水合反应对PEO 膜组成和结构的影响更大,尤其是在中性腐蚀介质中。
从表4 所示的各种化合物的热力学参数可知[31],Mg(OH)2比MgO 的标准摩尔生成自由能(ΔfGmϴ)更低,表明其具有更高的化学稳定性。因此,有研究指出[7,20,24],MgO 在中性NaCl 溶液中浸泡35 h 以上,即可完全转变为Mg(OH)2。由于Mg(OH)2较MgO 的摩尔体积更大,所以水合反应在一定程度上可以起到“封堵”膜层中微观孔隙的作用,进而延缓腐蚀的发生[7,20,24]。同时,相较于MgO,Mg(OH)2中O 的质量分数更大。因此,如图7c 所示,在PEO 膜层上尚未出现明显腐蚀痕迹的区域,并没有观察到如原始膜层结构中所见的大尺度孔隙,也没有观察到微裂纹,同时浸泡后的膜层中O 含量升高。
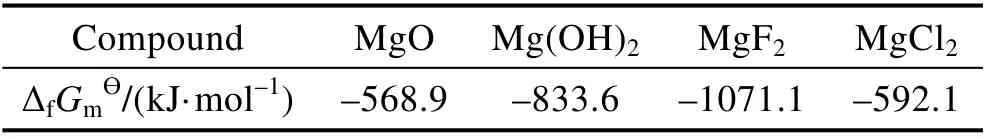
表4 膜层化合物与腐蚀产物的标准摩尔生成自由能Tab.4 The standard molar Gibbs free energy of formation of coating compounds and corrosion products
对于主要组成为MgF2的PEF 膜而言,水合反应直观的影响便是膜层中O 含量的增多。然而中性腐蚀介质对PEF 膜层组成的影响非常小,酸性腐蚀介质的影响相对较为显著,但在膜层中,F 的含量仍然远高于O。经历了中性介质的作用后,PEF 膜层的微观结构几乎没有发生变化;经历了酸性介质的作用后,膜层表面O 富集的颗粒物导致膜层中O 含量的增多,可能正是水合反应的产物[30]。从表4 中也可以看出,在热力学上,MgF2的化学稳定性远高于MgO和Mg(OH)2,表明PEF 膜难以发生此类水合反应。换言之,水合反应对于PEF 膜层的破坏作用较小。
除水合反应外,腐蚀介质中的Cl–部分占据MgO晶格位点,破坏水合产物Mg(OH)2,同时形成的可溶性化合物MgCl2也会破坏PEO 膜,这也是膜层发生腐蚀破坏的主要原因之一[20,24]。Cl–对膜层的溶解作用随着其浓度的上升而增强,这种溶解作用在膜层中的微裂纹处远甚于其他区域。动态电位极化测试中,施加外场迫使基材与腐蚀介质之间进行电荷交换,同样也会加剧Cl–的溶解作用及其在膜层厚度方向上的渗透作用。PEO 和PEF 膜中都没有贯通膜层的孔隙,但是PEO 膜层中孔隙的尺度更大,且含有较多的微裂纹,更有利于Cl–溶解作用和渗透作用的发生。
在酸性介质中,除了以上的水合反应与Cl–位点置换的溶解作用,还多了酸液对膜层化合物的消耗作用[8]。酸性浸泡实验结束后,两组膜层的截面形貌如图15 所示,图中“Resin2”为制备截面样品时的镶嵌树脂,“Resin1”为原始样品边缘处的封装保护树脂。从图15a 中可以直观地看出,PEF 在酸性浸泡实验前后,厚度并没有发生显著变化。相应的元素分布图表明,浸泡后,膜层中的O 含量有所增加,与图13a 中组成分析的结果一致。从图15b 中可以看出,酸性浸泡后,PEO 膜层消耗殆尽,在截面上仅观察到了一些残留的膜层碎片,结合O 的分布图还可以辨识出膜层内部位于膜基面处致密的阻挡层。由此可见,酸性腐蚀介质对于PEO 膜的溶解消耗非常严重,虽然对PEF 膜影响较小,但在一定程度上加剧了PEF膜的水化反应。
同时,由于腐蚀产物Mg(OH)2在酸中的溶解作用,在酸性浸泡实验中,膜层表面形成了裸露的腐蚀坑,没有堆积腐蚀产物。而没有腐蚀产物堆积形成的屏障,也加剧了酸性介质在膜层中的扩散和反应。

图15 两种膜层在酸性腐蚀介质中浸泡28 h 后的截面形貌Fig.15 Cross-sectional Morphologies of two coatings after immersion in acidic corrosive medium for 28 h: a) PEF coating; b)PEO coating
基于以上关于PEF 膜与PEO 膜在腐蚀行为上的对比分析,评价了PEF 膜层的腐蚀防护性能,并揭示了其腐蚀失效机制。在分析酸性浸泡后的PEF 膜层截面形貌时,获得了一组能够直观反映其腐蚀发生与发展过程的照片,如图16 所示。其中,图16a 为腐蚀介质穿透PEF 膜到达膜基面附近与基材发生了反应,形成的腐蚀产物(如方框所示)顶起了膜层,使膜层局部与基材脱离(如箭头所示);随着腐蚀的进行,腐蚀产物增多并在膜基界面附近堆积,如图16b 所示;由于腐蚀产物氢氧化镁的摩尔体积大于镁合金基材,导致局部膜层的断裂和脱落,如图16c 所示;失去膜层的区域,基材金属持续受到腐蚀介质的侵蚀,导致腐蚀区域在横向和纵向上都不断扩展,形成了宏观上的腐蚀坑,如图16d 所示。

图16 PEF 膜在酸性腐蚀介质中腐蚀的发生与发展过程Fig.16 Corrosion initiation and development in PEF coating in acidic corrosive medium
综上,对于PEF 膜的腐蚀防护失效机制而言,腐蚀介质在膜层中的扩散和渗透占据主导作用。对于PEO 膜的腐蚀防护失效机制而言,中性腐蚀介质以扩散和渗透作用为主,酸性介质则以溶解消耗为主。对于浸泡实验和盐雾实验而言,腐蚀防护失效的判据在于膜层表面出现腐蚀痕迹,一般是宏观上肉眼可以辨识的腐蚀点(坑)或是堆积的白色腐蚀产物。然而,腐蚀痕迹出现的时间显然滞后于腐蚀介质穿透膜层到达基材表面的时间,在这个时间间隔里,腐蚀介质与基材反应,且生成一定量的腐蚀产物,以至于“顶破”膜层(如图16 所示)。只有PEO 在酸性介质中的腐蚀过程例外,原因在于溶解消耗过程决定了其腐蚀机制。
电化学测试结果显示,PEF 膜的开路电位(OCP)和自腐蚀电位(Ecorr)都比PEO 膜高,表明其在热力学上更稳定,腐蚀倾向更低。这也得到了各组宏观腐蚀实验结果的印证。相应地,表4 中的热力学判据也表明,MgF2较MgO 的热力学稳定性更高。对于动态电位极化测试而言,自腐蚀电流密度(Jcorr)反映了膜层在自腐蚀电位(Ecorr)下发生腐蚀破坏的速率。该电流由腐蚀介质中的阴、阳离子穿越双电层(位于膜层与腐蚀介质的界面附近),在阳极表面(层)发生电荷交换所致。由于PEF 和PEO 膜的多孔结构及其亲水特性,这里的阳极表面(层)可能是膜层表面,也可能是在深至膜层与基材的界面附近。这种电荷交换的本质是,上述阳极表面(层)所发生的电极反应主要受电极状态的影响[32]。电化学理论指出[33],多孔电极比整体电极复杂得多,包含了多种传输过程,并且传输过程与电化学反应过程在相互交错的区域内同时进行。PEF 膜较PEO 膜的表面孔隙率更高,微孔数目更多且尺度更小,可见两组膜层与腐蚀介质界面的实际面积相差悬殊。从膜层微观形貌上来看,两组膜层在孔隙形状的规则程度与孔隙分布的均匀性上也存在很大的差异,并且在亲水性上也有很大的差异。因此,两组膜层所对应的电极状态非常复杂,其在电极反应机制上可能存在较大的差异。这也就导致电化学测试中,自腐蚀电流密度反映的腐蚀速率与宏观腐蚀过程难以对应,具体体现为,尽管PEF 膜的自腐蚀电流密度高于PEO 膜的自腐蚀电流密度,但却在浸泡实验和盐雾实验中表现出更为优异的腐蚀防护性能。
4 结论
1)相较于PEO 膜的多孔结构,PEF 膜具有孔隙率高,孔隙尺寸小,但孔隙数量多、形状不规则的特点。同时,PEF 膜表面和内部几乎没有微裂纹缺陷。
2)PEF 膜与PEO 膜均可以为镁合金基材提供有效的腐蚀防护作用。
3)PEO 膜在中性环境中的腐蚀防护失效机制主要是,腐蚀介质扩散穿透膜层,致使基材发生腐蚀;在酸性环境中的腐蚀防护失效机制主要是,膜层化合物的溶解和消耗。在中性和酸性环境中,PEF 膜的腐蚀防护失效机制主要都是,腐蚀介质扩散穿透膜层,致使基材发生腐蚀。
4)相较于PEO 膜,PEF 膜的腐蚀倾向更低,总体上腐蚀速率也更低,但在动态电位极化测试中的自腐蚀电流密度更高,可能与多孔电极上电极反应的复杂性有关。
