不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的电催化降解性能
2021-06-30唐静宜蔡毅雄邵艳群张燕斌
唐静宜,蔡毅雄,邵艳群,,张燕斌
(1.三峡大学电气工程及自动化学院,宜昌 443002;2.福州大学至诚学院材料工程系,福州 350002;3.福州大学材料科学与工程学院,福州 350116)
0 引 言
电极材料在电化学氧化处理工业污染废水中处于核心位置,决定着电催化氧化反应的速率、目标污染物的处理效率以及电化学工艺成本[1-3]。电极材料应既具有优异的催化活性,能够对电化学反应中参与的有机物进行活化,又具有良好的耐腐蚀性和较长的使用寿命[4-5]。目前,通过调节电极的电子结构来改变电催化活性是电催化领域的研究热点[6-7]。Ti/RuO2-SnO2电极在去除对苯顺二醇[8]、Ti/RuO2-ZrO2-SnO2电极在处理有机废水[9]、Ti/SnO2-Sb/PbO2氧化物涂层电极在降解10 mg·L-1甲基苯酚[10]方面都取得了较好的处理效果。作者制备了不同RuO2掺杂量的Ti/SnO2-NiO-RuO2电极,研究了其微观形貌、元素价态、表面活性、比表面积及电催化降解性能,为催化降解难处理有机废水提供参考。
1 试样制备与试验方法
试验材料为西北有色金属研究院生产的经喷砂处理的TA2钛板,尺寸为20 mm×20 mm×1.5 mm,采用超声清洗去除表面油污,在70 ℃、质量分数为10%的硫酸溶液中浸蚀2 h后用去离子水冲洗。分别将SnCl2、NiCl2、RuCl3·H2O溶于乙醇溶剂中配制成浓度为2 mol·L-1的溶液,然后添加物质的量分数分别为0,9.1%,16.7%,23.1%,28.6%的RuO2制成前驱体溶液。以锡的名义载量为1.4 mg·cm-2吸取前驱体溶液均匀涂覆在钛板上,置于500 ℃的马弗炉中保温10 min后取出,重复涂覆8次,第8次保温时间为2 h,得到Ti/SnO2-NiO-RuO2电极。
采用ZEISS Supra 55型扫描电镜(SEM)观察电极表面微观形貌,并用附带的Oxford X-MAX 50型能谱仪(EDS)测试微区成分。采用Thermo Scientific K-Alpha+型X射线光电子能谱仪(XPS)测试电极表面各元素的价态。采用Micromeritics 2460型比表面分析仪测试电极的比表面积。
采用三电极体系在AUTOLAB PGST-3020N型电化学工作站上进行电化学试验,Ti/SnO2-NiO-RuO2为工作电极,饱和甘汞电极(SCE)为参比电极,铂片为对电极,电解质为浓度0.5 mol·L-1的H2SO4溶液,测试电极的循环伏安(CV)曲线、电化学阻抗谱以及极化曲线。循环伏安曲线测试的电位窗口为0~1.2 V,扫描速率为10 mV·s-1。电化学阻抗谱测试的振幅为10 mV,频率为0.05~105Hz。极化曲线测试的电位范围为1.0~2.8 V。采用LAND-CT2001A型蓝电电池测试系统测试电极的强化寿命,根据HG/T 2471-2001,电流密度为500 mA·cm-2,硫酸溶液浓度为0.5 mol·L-1,当电解电压超过15 V时认为电极失活,此时电解反应累积的时间即为强化寿命。
将14.2 g Na2SO4和20 mg甲基橙(MO)溶于1 L蒸馏水中模拟工业废水,以Ti/SnO2-NiO-RuO2电极为工作电极,工作表面积为20 mm×20 mm,钛为辅助电极,在两电极体系下采用恒电流法对废水进行降解,降解电流为0.1 A。每隔0.5 h取一定量的溶液,采用Cary 50型紫外可见分光光度计测试紫外可见吸收(UV-Vis)光谱。采用TOC-L ASI-L型总有机碳分析仪测试降解不同时间后溶液的剩余有机碳含量。有机碳初始含量(质量浓度,下同)C0与剩余有机碳含量C之差,与初始含量的比值为降解率。
2 试验结果与讨论
2.1 微观形貌
由图1可以看出:RuO2掺杂量(物质的量分数,下同)为9.1%时,Ti/SnO2-NiO-RuO2电极表面随机分布许多细小的裂纹,此时涂层与基体的结合力较差;RuO2掺杂量为23.1%时,电极表面较平整,龟裂程度低,存在少量细小的浅裂纹,适量的浅裂纹能够为电催化过程提供更多的活性位点,并且有效增加电极的比表面积,使得有机物与电极表面充分接触而发生氧化降解;随着RuO2掺杂量进一步增加,电极表面变得光滑平整,涂层致密性得到改善。

图1 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的表面SEM形貌Fig.1 Surface SEM morphology of Ti/SnO2-NiO-RuO2 electrodes with different doping amounts of RuO2
由图2可以看出,RuO2掺杂量为23.1%时,Ti/SnO2-NiO-RuO2电极表面的锡、钌、镍、氧、氯等元素分布均匀,无钛元素,表明基体被涂层完全覆盖。由表1可以看出:不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极表面氧元素的原子分数均超过50%,表明前驱体溶液加热后转变成SnO2、NiO和RuO2氧化物的比例较高,锡、钌、镍元素大部分以氧化物的形式存在于涂层中。

表1 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极表面的EDS分析结果

图2 RuO2掺杂量为23.1%时Ti/SnO2-NiO-RuO2电极表面面扫区域和元素面分布Fig.2 Mapping region (a) and element mapping (b-f) on Ti/SnO2-NiO-RuO2 electrode surface with RuO2 doping amount of 23.1%
2.2 元素的组成和价态
电极的导电性与元素的价态密切相关,而元素的价态又与电子结构有关[11-13]。掺杂RuO2会影响元素的电子结构,从而影响电极的导电性。由图3(a)可以看出:Ru 3d核心能级谱是一对相对较窄的特征峰,对Ru 3d光谱的主自旋轨道双峰进行拟合,发现结合能为284.5 eV时出现的干扰信号峰C 1s与Ru 3d峰发生了部分重叠;RuO2掺杂量为9.1%时,位于Ru 3d5/2(281.4 eV)和Ru 3d3/2(284.6 eV)的峰对应为涂层热氧化得到的Ru4+;结合能为281 eV左右时还有Ru3+的峰;结合能为285.2 eV左右时,还存在RuOx峰,推测为高价(Ru6+)钌化物;与RuO2掺杂量为9.1%的电极相比,RuO2掺杂量为23.1%电极的Ru 3d3/2特征峰发生了红移,偏移量为0.25 eV。
由图3(b)可以看出,未掺杂RuO2电极的Ni 2p特征峰由结合能分别为855.1,872.6 eV的Ni 2p3/2和2个Ni 2p1/2自旋轨道耦合双峰以及结合能分别为862.4,879.9 eV的镍卫星峰组成,结合XPS数据库可知,Ni 2p特征光谱中镍元素呈现的价态分别为Ni2+和Ni3+,导致Ni 2p峰产生了蓝移现象。由表2可以看出,不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极中RuOx和Ru4+的占比均比Ru3+的大,Ni2+的占比比Ni3+的大得多,且随RuO2掺杂量增加,Ru4+的占比增大。在电极催化降解过程中,Ru4+起到氧化作用,Ni2+起到还原作用,二者的占比均较大,因此电催化过程掺杂RuO2电极的氧化还原作用较未掺杂RuO2电极的强。此外,Ru4+的掺杂会引起SnO2晶格畸变,造成各种离子之间电荷的不平衡,促使涂层表面生成更多的氧空位。氧空位可以通过促进对有机物的吸附来充当催化反应的活性位点,有利于提高电极的催化活性[11]。

表2 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极表面离子占比
由图3(c)可以看出:未掺杂RuO2电极的O 1s的2个信号峰的结合能分别为530.22,532 eV,分别代表涂层表面的晶格氧(Olatt)和化学吸附的活泼氧(Oads)。RuO2掺杂量分别为0,9.1%,23.1%时,Oads的物质的量分数分别为15.68%,37.42%,56.55%。Oads的含量会影响电极的电催化效果。

图3 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极表面的XPS光谱Fig.3 XPS spectra of Ti/SnO2-NiO-RuO2 electrode surface with different doping amounts of RuO2
由图3(d)可以看出,Sn 3d特征峰在486.2,494.8 eV的峰位均未发生移动,这是由于SnO2是一种化学状态较为稳定的半导体氧化物。
综上:掺杂RuO2后电极中离子的价态种类增多,电催化降解过程反应更活泼,有利于电催化降解的进行。
2.3 比表面积
电极的催化性能与其比表面积有关,比表面积越大,电催化性能越好。不同RuO2掺杂量的电极均具有介孔结构。由图4可以看出,随RuO2掺杂量增加,电极的总孔体积和比表面积均先增大后减小。结合XPS结果和SEM形貌可知:RuO2掺杂增大了电极表面的化学吸附氧浓度,增加了电极表面活性位点的数量,这说明比表面积增大;掺杂过量的RuO2时,电极的致密性提高,电极表面的氧化物涂层趋于封闭,因此掺杂过量RuO2时电极的比表面积反而降低。

图4 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的比表面积Fig.4 Specific surface areas of Ti/SnO2-NiO-RuO2 electrodes with different doping amounts of RuO2
2.4 电化学性能
2.4.1 交流阻抗
采用如图5(a)所示的等效电路对电极表面电荷转移和析氧过程的阻抗谱进行拟合,阻抗谱见图5(b),拟合参数见表3。图5(a)中:Rs为溶液电阻元件,其值为高频区曲线与x轴的交点;L为感抗元件,其值主要与电极的微观结构有关;Cdl为电极/电解质界面的双电层电容元件;Rf为电极/基体之间的电阻元件;Zw为Warburg阻抗元件,其值为曲线高频区的斜率;Rct为法拉第电荷传递电阻元件,其值为图5(c)中的半圆半径;Q为常相位角元件。由表3可以看出,随着RuO2掺杂量增加,电极/基体之间的电阻Rf、法拉第电荷传递电阻Rct均减小,双电极电容Cdl先增大后减小,这主要是由于晶态氧化钌优异的电荷传递特性能够显著降低电极的电荷转移电阻和涂层内阻,改善了电极的导电性,同时负载的氧化物之间(如RuO2、SnO2)的协同作用增强了电极的活性,使电极具有更加优异的催化性能。

图5 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的等效电路和交流阻抗谱 Fig.5 Equivalent circuit (a) and alternating current impedance spectra (b) of Ti/SnO2-NiO-RuO2 electrodes with different doping amounts of RuO2

表3 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的等效电路拟合参数
2.4.2 循环伏安曲线
由图6可以看出:不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的循环伏安曲线基本呈矩形,表明电极具有良好的氧化还原可逆性和电催化可逆性。在0.3~0.6 V电位间氧化还原峰的面积大小体现了电极活性位点的数量。RuO2掺杂量为23.1%时,氧化还原峰面积最大,说明电极活性位点数量最多,此时循环伏安曲线包围的面积最大,电极具有最高的伏安电荷密度,说明该电极的氧化还原作用最强,具有最好的电催化性能。
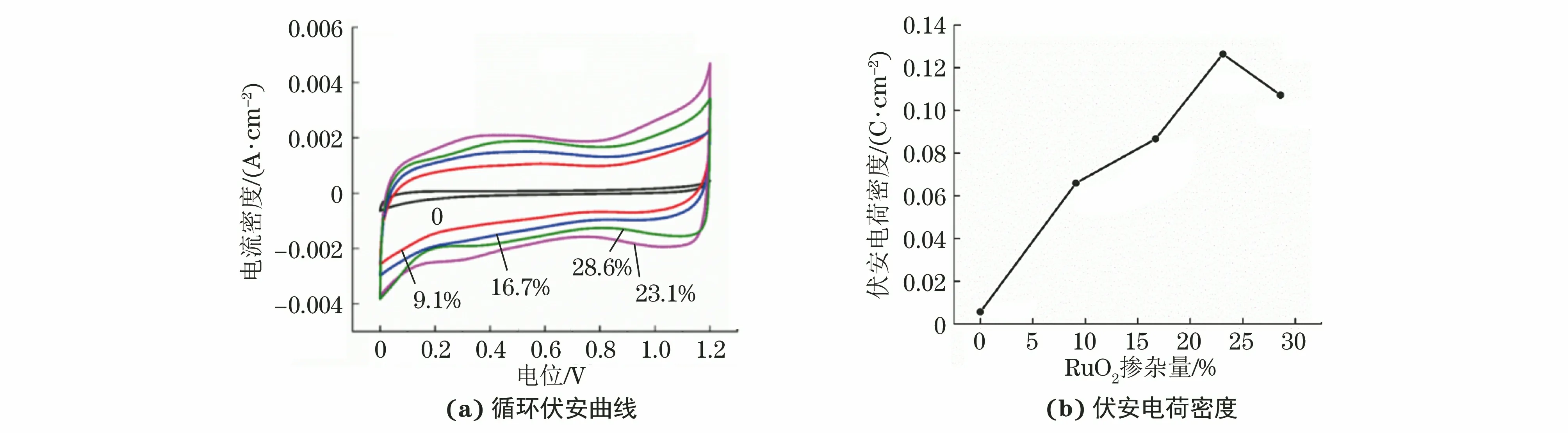
图6 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的循环伏安曲线和伏安电荷密度Fig.6 Cyclic voltammetric curves (a) and voltammetric charge density (b) of Ti/SnO2-NiO-RuO2 electrodes with different doping amounts of RuO2
2.4.3 极化曲线
由图7可以看出,不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的过电位均在1.6 V左右,比传统钌系阳极的析氧电位高。RuO2的掺杂使单位极板的电流密度增大,高的电流密度不仅能够加速电极表面各粒子的运动,使粒子与有机物充分接触并反应,还能促进电催化体系中主导氧化作用的·OH等活性自由基的生成。同时,RuO2的掺杂增强了电极的导电性和涂层表面对有机物的电化学吸附能力,加快了污染物在电极表面的氧化反应,有利于有机物的电催化降解。电流密度过高导致电极对偶氮类有机物的氧化分解能力下降,这主要是由于电流密度过高时体系中会发生阳极析氧副反应,在降解过程中产生电极自带的欧姆热阻效应,导致电流损耗,电极氧化分解污染物的效率也会明显降低[12]。
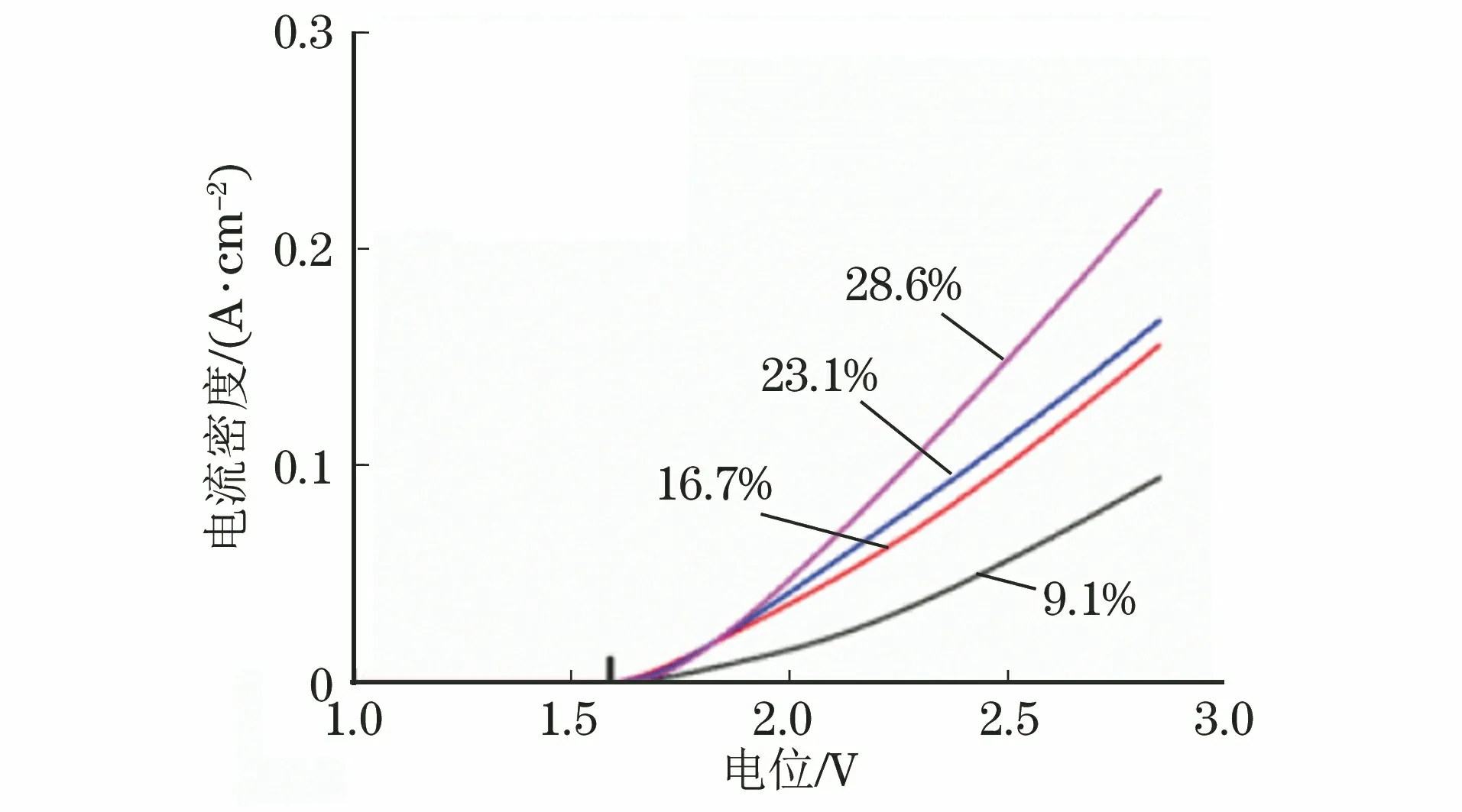
图7 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极的极化曲线Fig.7 Polarization curves of Ti/SnO2-NiO-RuO2 electrodes with different doping amounts of RuO2
2.4.4 强化寿命
由图8可看出,未掺杂RuO2电极的初始电位较高,随着RuO2掺杂量增多,初始电压降低,电极的强化寿命延长,在掺杂量为28.6%时达到最大,为582 h,较未掺杂RuO2电极的提高了十几倍。这主要是由于RuO2可以提高涂层的致密性,高温处理后,高结晶度的涂层与基体结合牢固,延长了电极的使用寿命。
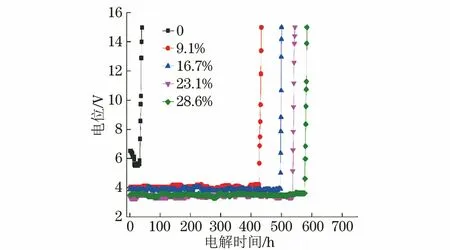
图8 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极在H2SO4溶液中的电位-电解时间关系曲线Fig.8 Potential-electrolytic time curves of Ti/SnO2-NiO-RuO2electrodes with different doping amounts of RuO2 in H2SO4 solution
2.5 电催化降解性能
由图9可以看出:随着电催化时间延长,MO吸收峰强度降低,吸光度迅速减小,且采用掺杂RuO2电极降解后的吸收峰强降低程度均明显高于采用未掺杂RuO2电极降解后的,说明RuO2有利于提高电极对MO溶液的电催化效果。

图9 采用不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极降解不同时间后MO溶液的UV-Vis光谱Fig.9 UV-Vis spectra of MO solution after degradation for different times by Ti/SnO2-NiO-RuO2 electrode with different doping amounts of RuO2
由图10可以看出:MO的降解速率随电催化时间的延长先增大后减小,这主要与电催化过程中有机物浓度越来越小以及形成的中间产物干扰有关;随着RuO2掺杂量增加,MO的降解率先增大后减小,掺杂量为23.1%时降解率最大,电极的催化效率最高,3 h后MO的降解率达87%,较未掺杂RuO2电极的提高了30%。这是由于适量RuO2的掺杂使涂层具有粗糙、均匀的表面形貌,高的比表面积、吸附氧浓度和伏安电荷量,提供了更多的电化学活性位点,使电极表面参与电催化反应的面积增大,增强了电极对污染物的吸附,促进了电催化。同时RuO2能提高电极材料的导电性,显著降低电极材料的电荷转移电阻和涂层内阻,提供了快速的电子转移通道,有利于反应快速进行。但是过多的RuO2会减小电极的比表面积,阻碍其对有机物的捕获,同时会在降解过程中产生过于剧烈的析氧副反应,减弱电极的电催化活性,阻碍降解反应的进行。

图10 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极电催化降解MO的降解率随降解时间的变化曲线Fig.10 Curves of degradation rate vs degradation time of MO degraded by Ti/SnO2-NiO-RuO2 electrodes with different doping amounts of RuO2
由图11可以看出,与未掺杂RuO2的电极相比,掺杂RuO2的电极能够更快地催化有机物降解,剩余有机碳含量更低,且随RuO2掺杂量增加,剩余有机碳含量先减少后增加。在掺杂量为23.1%时剩余有机碳含量最少,此时电极的电催化效果最好。在反应前1 h,50%(质量分数)左右的有机碳被快速去除,之后有机碳的含量下降平缓。这主要是由于降解前期的MO溶液初始浓度高,大量吸附在涂层表面并发生氧化分解和矿化。随着催化反应的进行,MO氧化分解形成的中间产物逐渐增加,需要更多的自由基团参与反应,其降解速率也随之减小。

图11 Ti/SnO2-NiO-RuO2电极电催化降解MO过程中剩余有机碳质量浓度的变化Fig.11 Variation of mass concentration of residual organiccarbon during degradation of MO by Ti/SnO2-NiO-RuO2 electrode
对剩余有机碳含量和降解时间进行拟合,关系式为
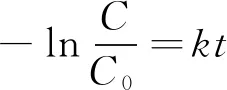
(1)
式中:k为一级动力学常数;t为降解时间。
k值反映了MO的降解速率。由图12可知,未掺杂RuO2电极对MO电催化降解的k值最小,k随RuO2的增加先增大后减小。虽然RuO2的掺杂有利于电极导电性的增大,但是过量掺杂会减少电极对有机物的吸附,减缓MO的氧化分解。RuO2掺杂量为23.1%时k值最大,说明此时MO的降解速率最快,这与前面的结果一致。

图12 不同RuO2掺杂量Ti/SnO2-NiO-RuO2电极电催化降解MO过程的动力学曲线Fig.12 Kinetics curves during electrocatalytic degradation of MO by Ti/SnO2-NiO-RuO2 electrodes with different amounts of RuO2
2.6 电催化降解机理


(2)

(3)
2·HO2→O2+H2O2
(4)
O2+2H++2e→H2O2
(5)

(6)
(7)
有机物从溶液迁移至Ti/SnO2-NiO-RuO2电极表面后发生电化学反应。其中·OH与电极表面的活性氧化物快速发生氧化反应,形成含有活性自由基·OH的金属氧化物RuOx[·OH]和高价态的金属氧化物RuOx+1(x>1)等媒介物质。O 1s光电子能谱分析表明,RuO2掺杂量为23.1%的电极含有的Oads的物质的量分数比未掺杂电极的高41%。金属氧化物阳极上的Oads是形成高价态媒介物质(RuOx+1)的关键,Oads浓度越高,RuOx+1越多。RuOx[·OH]参与有机物降解,而RuOx+1的含量决定电催化的速率。掺杂RuO2电极的比表面积和吸附氧含量较高,导致活性自由基增加,形成更多高价并对有机污染物有较好选择性的强氧化物RuOx+1,加上电极导电性增强和活性位点增多,反应过程中电子转移速率加快,在电化学的作用下可以更快地进行氧化还原反应,提高对目标产物的降解速率。同时,高价态的氧化物在降解过程中重新被还原至初始价态。催化反应过程不断循环,从而去除废水中的污染物[13-14]。
RuO2+·OH→RuO2[·OH]
(8)
RuO2[·OH]+R→R[·OH]+RuO2
(9)
RuO2[·OH]→RuO2-x+RuOx+1+H++e
(10)
RuOx+1+2R[·OH]→2RO+RuOx+H2O
(11)
3 结 论
(1) 掺杂RuO2可以增加Ti/SnO2-NiO-RuO2电极表面的吸附氧浓度、活性位点数量、离子的价态种类,提高电极的比表面积、伏安电荷密度,促进电催化降解的进行,同时还能延长电极的强化寿命。
(2) 当RuO2掺杂量为23.1%时,Ti/SnO2-NiO-RuO2电极对甲基橙的电催化降解速率最快,降解率最大,3 h后甲基橙的降解率达到87%,较未掺杂RuO2电极的提高了30%,该电极具有良好的电催化效果。
