水稻干旱胁迫的small RNA转录组分析
2021-06-28刘源张秀妍徐妙云郑红艳邹俊杰张兰王磊
刘源, 张秀妍, 徐妙云, 郑红艳, 邹俊杰, 张兰, 王磊
(中国农业科学院生物技术研究所, 北京 100081)
干旱胁迫影响植物生长发育、生理代谢、地理分布、品质产量,是威胁粮食安全的主要环境因素之一[1]。在干旱条件下,植物会产生一系列结构和生理改变,如叶片和根系形态改变、气孔运动、渗透损伤、物质吸收失衡、光合能力降低等[2-5]。植物在干旱响应过程中,大量抗旱相关调控基因的表达水平会发生改变,包括转录因子基因、渗透调节基因、抗氧化代谢基因和逆境诱导基因等,其中,许多基因在转录和转录后水平受small RNA调控[2, 5]。因此,small RNA及其靶基因组成调控模块是植物干旱胁迫响应过程中的关键因子。
microRNA(miRNA)是广泛分布于植物基因组中的内源性单链非编码small RNA,长度18~24个核苷酸(nt)。miRNA通过结合靶基因上的互补序列诱导转录水平基因沉默(transcriptional gene silencing,TGS)和转录后水平基因沉默(post-transcriptional gene silencing,PTGS)而发挥功能[6]。miRNA不仅是植物生长发育和生理代谢活动的重要调节因子,同时也参与了各种生物和环境胁迫(干旱、高盐、高温、冷冻等)的响应过程[6-7]。目前,许多植物物种的干旱胁迫紧密相关或响应miRNAs已被鉴定和研究[2-4]。然而,干旱逆境适应和响应是一个非常复杂的过程,更多干旱调控关键miRNAs及其功能有待鉴定和解析,而且miRNA-靶基因调控干旱胁迫的分子机制仍不完全清楚。
本研究拟通过高通量RNA-seq技术对水稻根、茎和叶片中small RNA在干旱处理后全基因组水平上的表达变化情况进行研究,鉴定已知miRNAs和新miRNAs、筛选和挖掘干旱响应差异表达miRNAs并对其预测的调控靶基因进行功能注释和富集分析,为进一步探明水稻干旱胁迫应答和适应过程中的miRNAs调控机制及其分子调控通路和网络提供理论和数据参考。
1 材料与方法
1.1 水稻材料培养
水稻材料OsPIR1-RNAi(背景为OryzasativaL. ssp.japonica)最初来自本课题组构建的RNAi突变体库[8],为OsPIR1基因(Os03g0845000)的RNA干扰(RNA interference)植株。首先,选取该材料T2代的种子经75%乙醇和2.5% NaClO(体积分数)消毒后在滤纸上室温萌发约48 h,然后转移至土壤,在16 h光照/8 h黑暗,光照强度约8 000 lx,28 ℃光照培养箱生长至幼苗二叶期。
1.2 干旱处理与样品收集
选取二叶期生长相对一致的水稻植株转移至中国农业科学院生物技术所温室内继续培养,从该时期的水稻幼苗开始持续断水处理。处理0、24、96 h采集根(root,R)、茎(stem,S)及叶(leaf,L),分别命名为R-0 h、S-0 h、L-0 h、R-24 h、S-24 h、L-24 h、R-96 h、S-96 h、L-96 h,共9个样品,立即置于液氮中保存备用。
1.3 Small RNA文库构建及测序
采用EASYspin Plus多糖多酚复杂植物RNA快速提取试剂盒(RN5301,Aidlab)提取9个样品的总 RNAs,通过反转录合成cDNA第一链。PCR扩增后经过纯化和大小选择构建small RNA文库并进行质量评估[9-10]。将构建合格的文库使用北京贝瑞和康生物技术有限公司Illumia Solexa测序平台进行高通量single-end测序。
1.4 测序数据质量控制及注释
首先将Illumia测序得到的图像经碱基识别[11]转化为FASTQ格式的原始测序数据(raw reads)。去除原始数据中低质量读数、接头序列、冗余片段及短于18或长于30个核苷酸的序列,获得高质量序列(clean reads),统计测序碱基质量值Q30等[11-12],尽可能地保证测序结果的准确性。将clean reads利用Bowtie软件(v1.0.0)[13]比对Silva(http://www.arb-silva.de/)、GtRNAdb(http://lowelab.ucsc.edu/GtRNAdb/)、Rfam(http://rfam.xfam.org/)和Repbase(http://www.girinst.org/repbase/)等数据库进行注释,包括核糖体RNA(rRNA)、核内小RNA(snRNA)、核仁小RNA(snoRNA)、转运RNA(tRNA)等非编码RNA以及重复序列(repbase),未比对上的clean reads则被认定为未注释序列(unannotated reads)。
1.5 已知miRNAs和新miRNAs鉴定
使用Bowtie软件[13]将未注释序列与水稻参考基因组(Oryza_sativa.IRGSP-1.0)进行序列比对,获取其在基因组上的位置信息。将比对到参考基因组的序列(mapped reads)与miRNA数据库miRBase(release 22;http://www.mirbase.org/)中的已知成熟miRNA序列及其上游2 nt与下游5 nt范围内进行比对,最多允许一个碱基错配,能比对到的序列即被鉴定为已知miRNAs。利用miRDeep2软件(v2.0.5)[14],通过mapped reads在参考基因组上的位置信息得到潜在前体序列,基于miRNA前体特征二级发夹结构及前体结构能量信息(RNAfold randfold)采用贝叶斯模型经打分最终实现新miRNAs的预测[15]。
1.6 差异表达miRNAs分析
首先将所有样本中miRNAs表达量采用TPM(transcript per million)值进行标准化或归一化处理[16]。将干旱处理后24 和96 h的根茎叶(R/S/L-24 h、R/S/L-96 h)分别与0 h对应的组织(R/S/L-0 h)进行两两比较,使用edgeR软件(v3.8.6)[17]用来确定差异表达,以|log2(FC)|≥1.0(fold change, FC)和FDR≤0.1(false discovery rate, FDR)为标准筛选差异表达的miRNAs。利用在线平台(http://bioinformatics.psb.ugent.be/webtools/Venn/)构建韦恩图。使用R packages(http://www.rproject.org/)对差异表达的miRNAs以log2(TPM+1)值进行层次聚类分析,TPM (transcripts per million)为标准化的miRNA表达值。
1.7 靶基因预测及功能注释
利用TargetFinder平台(v1.6)[18-19]对获得的差异表达的已知miRNAs和新miRNAs靶基因进行预测。然后通过BLAST软件将靶基因序列与NR(ftp://ftp.ncbi.nih.gov/blast/db/)、Swiss-Prot(http://www.uniprot.org/)、GO (http://www.geneontology.org/)、COG(http://www.ncbi.nlm.nih.gov/COG/)、KEGG(http://www.genome.jp/kegg/)、KOG (ftp://ftp.ncbi.nih.gov/pub/COG/KOG/)、Pfam(http://pfam.xfam.org/)等数据库进行检索比对,进而对靶基因进行功能注释。
1.8 靶基因GO与KEGG富集分析
使用GOseq[20]和KOBAS[21]对靶基因进行GO富集分析(Wallenius non-central hyper-geometric distribution)和KEGG富集分析。
2 结果与分析
2.1 水稻干旱处理后small RNA的数量变化
干旱处理0、24和96 h的水稻根组织分别获得4 120 407、2 538 241和2 130 755条高质量序列(clean reads);茎组织分别产生2 506 166、1 371 020和1 407 041条高质量序列;叶组织分别产生2 995 912、1 741 575和1 617 673条高质量序列。通过质量控制,每个样本碱基质量值Q30均大于94%,表明small RNA-seq建库测序过程中碱基识别精度很高,数据质量可靠。
干旱处理后0、24和96 h的水稻根组织产生的未注释序列(unannotated reads)占所有small RNA比例分别为87.15%、72.19%和76.57%;茎的占比分别为90.4%、71.78%和77.48%;叶的占比分别为92.06%、69.98%和79.34%(表1)。将以上得到的unannotated reads映射到水稻参考基因组,能比对到基因组正链(+)或负链(-)上的即为mapped reads。干旱处理0、24和96 h的水稻根组织mapped reads占所有unannotated reads比例分别为55.75%、13.42%和21.04%;茎的占比分别为60.05%、50.77%和43.78%;叶的占比分别为53.18%、39.04%和46.05%(表1)。这些结果表明,干旱处理后水稻根、茎和叶组织中的clean reads、unannotated reads、mapped reads均存在不同程度的下降。

表1 水稻small RNA分类注释和比对到水稻参考基因组信息统计Table 1 Summary of classification of rice small RNA and unannotated reads mapped on rice genome
2.2 水稻干旱处理后small RNA的长度分布变化
small RNA的长度一般为18~30 nt,其中21~24 nt的small RNA在植物体内丰度较高且发挥了重要的调控作用。对干旱处理后水稻根、茎和叶组织中small RNA长度分布的变化趋势进行分析,结果(图1)表明:①R/S/L-0 h、S/L-24 h和R/S/L-96 h中24 nt small RNA丰度最高,而R-24 h中21 nt small RNA丰度最高;②随着干旱处理时间的延长,水稻根和茎中21 nt small RNA丰度先升高后下降,而叶片中的21 nt small RNA则不断下降;③根、茎和叶中22 nt small RNA丰度在处理后均呈现持续下降趋势;④随着干旱处理时间的增加,根中23 nt small RNA丰度先迅速下降后缓慢上升,而茎和叶中23 nt small RNA丰度则不断下降;⑤根、茎和叶中24 nt small RNA的丰度在干旱处理后均呈先下降后上升趋势。

图1 干旱处理后水稻根、茎和叶中small RNA长度分布Fig.1 Length distribution of small RNAs in rice root, stem and leaf under drought
2.3 水稻干旱处理后miRNAs变化
将mapped reads与miRBase数据库进行序列比对和茎环结构预测,结果(表2)发现,所有样品共鉴定到403个miRNAs,其中已知miRNA 352个,新预测miRNA 51个,而且水稻干旱处理后不同时间的根、茎和叶组织中已知miRNAs和新miRNAs表达数量不同。

表2 miRNAs数量统计Table 2 Summary of miRNAs number
2.4 水稻干旱处理后差异表达的miRNAs
根据表达量计算各miRNA在同一组织不同时间点样本间的表达水平变化情况,结果(表3)显示,水稻叶片干旱处理96 h后差异表达的miRNAs最多,而根部处理96 h后和茎部处理24 h后差异表达的miRNAs最少。其中,miRNAs表达上调数量最多的是干旱处理后96 h的叶,数量最少的是处理后24 h的根和茎;miRNAs表达下调数量最多的是干旱处理后96 h的茎部,数量最少的是处理后96 h的根部。为深入挖掘参与调控水稻干旱胁迫响应过程中的miRNAs,进一步对差异表达miRNAs在干旱处理后不同组织的变化趋势进行详细分析。
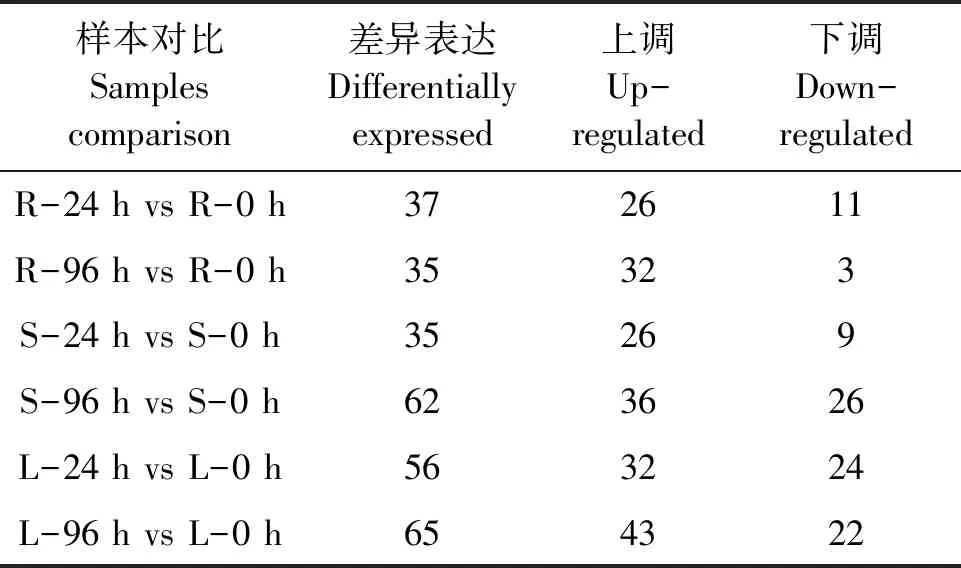
表3 干旱处理后水稻根、茎和叶差异表达的miRNAs数量Table 3 Number of differentially expressed miRNAs in drought-treated root, stem and leaf
2.4.1根差异表达miRNAs分析 从图2可以看出,21个miRNAs在处理后两个时间点的水稻根组织中都差异表达。根据表达量变化趋势,将这21个差异表达miRNAs分为三类。第Ⅰ类包含Osa-miR11337-5p和Osa-miR11340-5p,它们在干旱处理24 h内表达量迅速降低,在24 至96 h内逐渐升高;第Ⅱ类包含12个miRNAs,它们的表达量在干旱处理96 h内持续升高;第Ⅲ类包含7个miRNAs,它们的表达量在干旱处理24 h内迅速升高,在24 至96 h内逐渐下降。

图2 干旱处理后水稻根中差异表达miRNAs情况Fig.2 Changes in miRNAs expression of root under drought
对这21个差异表达miRNAs进行分析发现,Osa-miR166家族和Osa-miR820家族中表达发生变化的成员最多,其中Osa-miR166家族的4个成员表达量呈现先升高后下降趋势,而Osa-miR820家族的3个成员表达则持续升高,表明这两个miRNA家族可能在水稻根对干旱胁迫应答反应过程中发挥重要功能。
2.4.2茎差异表达miRNAs分析 从图3可以看出,28个miRNAs在处理后两个时间点的水稻茎组织中都呈现差异表达。根据表达水平变化趋势,将这28个差异表达miRNAs分为四类。第Ⅰ类包含4个miRNAs:Osa-miR167i-3p、Osa-miR408-5p、Osa-miR2878-5p和Osa-miR11340-5p,它们的表达量在干旱处理96 h内持续下调;第Ⅱ类包含3个miRNAs:Osa-miR1425-5p、Osa-miR5788和Osa-miR11337-5p,它们的表达量在24 h内迅速降低,在24 至96 h内逐渐升高;第Ⅲ类包含10个miRNAs,它们在干旱处理96 h内表达量持续升高;第Ⅳ类包含了11个miRNAs,它们的表达量在96 h内呈现先升高后降低的趋势。
差异表达的miRNA家族中,Osa-miR166家族的5个成员和Osa-miR159家族的2个成员在干旱处理后表达量呈现先升高后下降趋势,而3个新miRNAsnovel_miR_13、novel_miR_24和novel_miR_29则受干旱胁迫诱导持续上调表达,推断它们可能也参与了水稻茎部干旱响应分子调控网络。
2.4.3叶差异表达miRNAs的变化 从图4可以看出, 38个miRNAs在处理后两个时间点的水稻叶组织中都呈现差异表达。根据表达量变化趋势,将这38个差异表达miRNAs分为五类。第Ⅰ类包含8个miRNAs,它们的表达水平在干旱处理96 h内持续下降;第Ⅱ类包含4个miRNAs,它们在24 h内表达量迅速降低,在24 至96 h内逐渐升高;第Ⅲ类包含19个miRNAs,它们在干旱处理96 h内表达量持续上调;第Ⅳ类包含5个miRNAs,它们的表达量在96 h内先升高后降低;第Ⅴ类包含Osa-miR1425-3p和Osa-miR11342-5p,它们只在0 h的叶中表达,而在处理后24 和96 h均未检测到表达。
对这38个差异表达miRNAs进行进一步家族分析发现,Osa-miR160家族的3个成员、Osa-miR166家族的5个成员和Osa-miR820家族的3个成员表达量受干旱诱导持续上升,而Osa-miR159家族的2个成员表达水平随着处理时间的增加呈现先升高后降低趋势,表明这些miRNA家族在水稻叶片中响应干旱胁迫。
2.4.4根、茎和叶中共同差异表达miRNAs的变化 进一步对21、28和38个分别在水稻根、茎和叶差异表达的miRNAs进行分析,结果(图5)发现,共有15个miRNAs在干旱处理后24和96 h的各组织中均差异表达。其中Osa-miR159f、Osa-miR164f、Osa-miR5082和Osa-miR5493的表达水平随处理时间的增加而持续升高,表明它们受干旱胁迫诱导。因此,这些miRNAs是水稻营养组织干旱逆境适应与响应机制中关键调控因子。进一步对比水稻不同组织的差异表达miRNAs发现:Novel_miR_37在0 h的根、茎和叶中表达水平很低,而干旱处理后迅速上升;Osa-miR11337-5p和Osa-miR11340-5p在0 h的根、茎和叶中表达量较高,而处理后迅速降至很低水平;Osa-miR166c-3p在根、茎和叶中的表达变化趋势相同,均表现为先升后降;Osa-miR1876在根和叶中表达量随处理时间持续上升,而在茎中则先显著上调后缓慢下降。这些结果说明,不同处理时间的根、茎和叶组织具有明显不同的miRNAs表达谱,而且同一个miRNA在不同组织部位的表达水平也并不相同。

图5 干旱处理后水稻根、茎、叶共同差异表达的miRNAsFig.5 Differentially expressed miRNAs all in root, stem and leaf under drought treatment
2.5 预测的miRNAs靶基因及GO/KEGG富集
miRNA是通过调节靶基因的表达水平从而发挥生理功能,靶基因被进一步利用TargetFinder平台进行预测,最终发现403个miRNAs共对应4 537个靶基因。其中,352个已知miRNAs共对应3 883个靶基因,51个新miRNAs共对应785个靶基因。进一步使用数据库比对方法获得靶基因的详细功能注释信息,4 537个靶基因中4 484个获得注释信息。在干旱处理24和96 h后,根中差异表达的51个miRNAs共对应923个靶基因,茎中差异表达的69个miRNAs共对应641个靶基因,叶中差异表达的83个miRNAs共对应1 247个靶基因。
为了进一步了解干旱处理后水稻根、茎和叶组织中差异表达miRNAs对应的靶基因主要富集到哪些代谢途径和生物学过程,进行了GO和KEGG富集分析。结果(图6)表明:对于根,在GO生物学过程中靶基因显著富集于氧化还原、木质素分解代谢等过程,在GO细胞组分中显著富集于细胞膜相关囊泡和质外体等;在GO分子功能中显著富集于电子载体活性和血红素结合等,而角质、木栓质和蜡质生物合成及碱基切除修复等是显著富集的KEGG通路;对于茎,在GO分子功能中显著富集于ADP结合和染色质结合等,在GO生物学过程中靶基因显著富集于防御应答和细胞凋亡过程等,在GO细胞组分中显著富集于核仁组织区和叶绿体基质类囊体等,而甘氨酸、丝氨酸和苏氨酸代谢及脂肪酸生物合成等是显著富集的KEGG通路;对于叶,在GO生物学过程中靶基因显著富集于氧化还原和细胞凋亡过程等,在GO分子功能中显著富集于染色质结合和质子反向转运活性等,在GO细胞组分中显著富集于泛素连接酶复合体和细胞膜相关囊泡等,而角质、木栓质和蜡质生物合成以及油菜素内酯生物合成等是显著富集的KEGG通路。总之,这些结果为揭示水稻根、茎和叶组织响应干旱胁迫应答的分子细胞机制提供了线索和参考。

A:根;B:茎;C:叶;左图:GO分析;右图:KEGG富集分析。A: Root; B: Stem: C: Leaf; Left: GO analysis; Right: KEGG analysis.图6 干旱处理条件下差异表达miRNAs对应靶基因GO和KEGG分析Fig.6 GO and KEGG analysis of genes targeted by differentially expressed miRNAs after drought stress
3 讨论
miRNA不仅受干旱胁迫诱导而改变表达水平,也通过在转录和转录后水平调节干旱响应靶基因表达而参与植物干旱环境适应及干旱抗性应答过程[2, 5]。目前,已有研究使用芯片技术对断水干旱处理条件下分蘖期和花序形成期的水稻植株[22],以及利用small RNA测序对20% PEG模拟干旱胁迫条件下幼苗期的水稻根和苗组织[23]和断水干旱处理条件下开花期的水稻剑叶、穗[24]与成熟花序[25]进行了表达谱分析。本研究通过分析全基因组范围内miRNA转录组的变化深入挖掘干旱逆境相关miRNAs,并结合先前研究结果解析水稻不同组织响应干旱胁迫的调控网络。
miR159受干旱胁迫诱导而上调表达,而且它通过抑制MYB类靶基因的表达来参与ABA信号转导途径进而在干旱响应过程中发挥重要功能[3, 26]。本研究结果显示,Osa-miR159f的表达水平在营养组织中受干旱胁迫诱导持续上调,而Osa-miR159f的预测靶基因Os01g0812000、Os03g0578900、Os05g0490600和Os06g0605600编码MYB类转录因子,其中Os03g0578900和Os06g0605600已被报道在干旱条件下的水稻叶和花序组织中表达下调[27-28],表明干旱胁迫下Osa-miR159f与Os03g0578900和Os06g0605600可能存在负调控关系。miR164通过调节NAC类靶基因的表达水平来参与生长素信号并负调控干旱抗性[29-30]。本研究中Osa-miR164f在干旱处理后持续上调表达,而其预测靶基因Os02g0579000、Os04g0460600、Os06g0675600、Os08g0200600和Os12g0610600编码NAC类转录因子,其中Os02g0579000和Os08g0200600的表达在水稻叶或幼苗中受干旱诱导下调[27, 31],表明Osa-miR164f在干旱响应过程中可能负调控Os02g0579000和Os08g0200600。已有研究表明,miR166通过转录后调控Homeobox-leucinezipper(HD-ZIP)类靶基因的表达参与干旱胁迫下侧根发育[2, 4]。本研究中五个miR166家族成员在干旱处理后的营养组织中都差异表达,而它们的预测靶基因编码HD-ZIP类转录因子,其中Os12g0612700在干旱处理后的水稻叶中表达被下调[27],表明在干旱响应过程中它们可能也存在负调控关系。因此上述调控模块在水稻营养组织响应和抵御干旱胁迫过程中可能发挥关键作用。miR160、miR167、miR408和miR528也被证实涉及响应、缓解和抵御植物干旱逆境胁迫[2-4],而本研究中这些miRNAs也在干旱处理后根、茎或叶组织中也差异表达。Osa-miR5082和Osa-miR5493在营养组织中受干旱诱导显著上调表达,但它们在干旱胁迫调控网络中的具体功能目前尚未被报道。而且本研究中发现的许多差异表达的新miRNAs可能与干旱胁迫调控紧密相关,它们的具体分子机制也有待于进一步挖掘和解析。
植物在遭遇干旱胁迫时会通过干旱响应miRNAs-靶基因在分子水平启动干旱适应机制,进而改变生理代谢活动和形态结构如抗氧化物和渗透调节物质积累、细胞失水保护物质合成等来缓解或抵御干旱逆境[2,5,32-34]。本研究中差异miRNAs对应的靶基因显著富集在氧化还原途径、脯氨酸和α-亚麻酸(alpha-linolenic acid)代谢以及角质、木栓质和蜡质生物合成等,证实这些生物过程和代谢途径可能参与干旱逆境响应,但它们的具体作用机制仍待进一步探究。
总之,miRNAs在水稻根、茎和叶组织对干旱胁迫的适应和响应过程中扮演了重要的角色,而且本文构建的miRNAs-靶基因的调控模块也为进一步阐明和完善植物在干旱胁迫条件下结构、生理和分子改变的详细机理提供了理论基础,并将为分子遗传育种改良水稻抗旱性提供了分子数据资源。
