噬血细胞淋巴组织细胞增生症为首发的儿童非霍奇金淋巴瘤及淋巴细胞增殖性疾病6例
2021-06-22尹楠戈沈亚莉肖剑文2
尹楠戈,沈亚莉,朱 进,肖剑文2,△
1.重庆医科大学附属儿童医院药剂科,重庆 400014;2.儿童发育疾病研究教育部重点实验室,重庆 400014;3.儿科学重庆市重点实验室,重庆 400014;4.重庆医科大学附属儿童医院血液科,重庆 400014;5.重庆医科大学病理教研室,重庆 400016
噬血细胞淋巴组织细胞增生症(HLH)简称为噬血细胞综合征,是一组由活化的淋巴细胞和组织细胞过度增生,但免疫应答无效引起的多器官高炎性反应的临床综合征,主要表现为持续发热、脾大、血细胞减少和组织细胞噬血现象[1-2]。既往认为淋巴瘤尤其是非霍奇金淋巴瘤(NHL)、淋巴细胞增殖性疾病(LPD)相关HLH(LAH)成人较多而儿童期较少见[3-4],但随着对本病的重视和研究的深入,儿童期LAH的报道也逐渐增加。HLH起病急且进展快,早期病理检查往往难以进行,因此,LAH的早期诊断和及时治疗均较困难。重庆医科大学附属儿童医院血液科近期收治6例以HLH起病的儿童NHL或LPD,现将其临床特征和诊治情况报道如下。
1 资料与方法
1.1一般资料 2018年4月至2019年3月重庆医科大学附属儿童医院血液科收治初诊HLH患儿57例,其中6例以HLH为首发表现并最终确诊为NHL或LPD,收集并回顾性分析6例患儿资料,包括其临床特点及实验室检查结果、治疗及预后情况等。
1.2诊断标准 HLH诊断均符合HLH-04标准[5],即发现HLH相关分子遗传学异常或满足下列标准8条中的至少5条:(1)持续发热超过7 d;(2)脾大;(3)两系以上的血细胞减少;(4)三酰甘油(TG)≥3 mmol/L和(或)纤维蛋白原≤1.5 g/L;(5)骨髓、脾脏或淋巴结检查发现噬血细胞;(6)NK细胞活性降低(正常值≥15.11%);(7)血清铁蛋白≥500 mg/L;(8)可溶性CD25(sCD25)≥2 400 U/mL。NHL及LPD的诊断均依据WHO 2016年修订的造血及淋巴组织恶性肿瘤诊断标准[6](WHO-2016标准)完善病理诊断及分型。所有患儿均接受全身增强CT或PET-CT检查明确有无淋巴结肿大、肝脾大及其他结外部位受累。
1.3治疗方案及疗效评估 若明确为NHL或LPD,则根据病理类型采用不同的化疗方案。HLH疗效评估参照噬血细胞性淋巴组织细胞增生症诊疗建议[7]分为有效、疾病缓解、疾病活动及疾病复发;NHL及LPD的疗效评估则参照儿童非霍奇金淋巴瘤诊疗建议(NHL-09方案)[8]分为完全缓解(CR)、部分缓解(PR)、无进展及进展。

2 结 果
2.1临床资料 6例患儿包括2例男性和4例女性,起病年龄37~210月,有4例发病年龄>5岁。起病时6例均持续高热及脾大,5例淋巴结肿大,其中颈淋巴结肿大2例、PET-CT或增强CT提示腹腔淋巴结稍大3例。6例均全血细胞减少、铁蛋白增加及骨髓有噬血现象,骨髓流式细胞术(FCM)检查未见明显免疫表型异常细胞,sCD25>44 000 U/mL。6例患儿中,5例铁蛋白>1 000 mg/L,4例乳酸脱氢酶(LDH)≥正常上限3倍,4例纤维蛋白原<1.5 g/L、3例TG≥3 mmol/L及NK细胞比例低于正常值。5例患儿进行了NK细胞活性检测,2例低于正常值。见表1。

表1 初始临床资料

病例NK细胞活性铁蛋白(mg/L)sCD25(U/mL)血EBV-PCR(copy/mL)脑脊液EBV-PCR(copy/mL)LDH(U/mL)117.84%1 578>44 0006.36×1041.89×1074 055212.12%1 919>44 0001.06×10501 726310.14%3 155>44 0003.97×107ND966417.52%641>44 00002.85×107784511.48%1 344>44 0006.42×1061.73×1041 3236ND1 134>44 0002.48×1051.40×104555
2.2治疗及预后 1例患儿初诊时无明显淋巴结肿大,3例患儿腹腔淋巴结肿大但病情重故未做活检,上述4例按照HLH-04方案治疗2~8周均CR,但再次表现为HLH且颈淋巴结肿大,活检确诊NHL及LPD各2例。2例NHL中,EB病毒(EBV)阳性(EBV+)弥漫性大B细胞淋巴瘤(DLBCL)和高增殖性的B细胞非霍奇金淋巴瘤(B-NHL)各1例,分别按照NHL-09方案[8]+美罗华、BFM-95方案[9]+美罗华化疗达CR,均已停药。2例LPD均为系统性EBV+LPD,其中T细胞LPD(T-LPD)及B细胞LPD(B-LPD)各1例,B-LPD患儿给予BFM-95方案1个疗程达到CR,放弃治疗2周后死亡;T-LPD患儿放弃治疗2周后死亡。见表2。其余2例患儿初诊即颈淋巴结肿大,活检确诊ALK阳性间变性大细胞淋巴瘤(ALCL)及EBV+T-LPD各1例。ALCL患儿接受NHL-09方案化疗CR,已停药;EBV+T-LPD患儿接受甲强龙+足叶乙苷化疗2周达到PR,后因疾病进展死亡。随访至2020-06-30,3例EBV+LPD死亡,3例NHL生存。NHL患儿与EBV+LPD患儿的OS分别为(24.15±3.09)个月与(2.66±0.96)个月,NHL患儿生存时间明显长于EBV+LPD患儿,差异有统计学意义(P=0.024 6)。
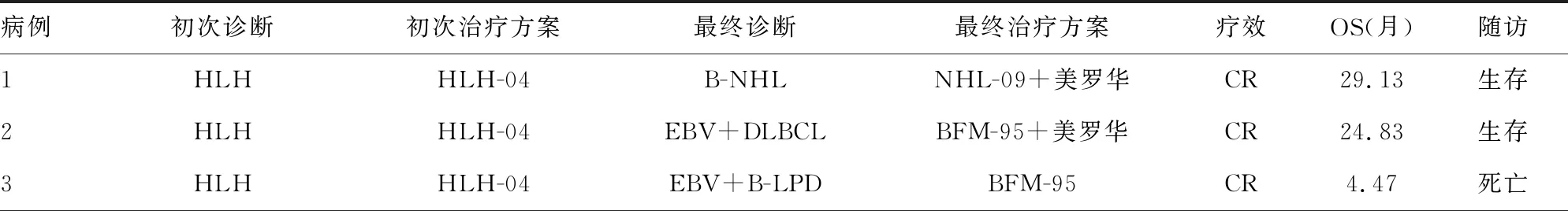
表2 治疗、诊断及转归

续表2 治疗、诊断及转归
2.3二代测序结果 共5例患儿利用二代测序(NGS)技术完成全外显子组(WES)测序[10]。4例患儿分别检出了1~6种不同类型的致病基因,B-NHL及EBV-B-LPD患儿各1例检出遗传性肿瘤易感基因,见表3。

表3 全外显子组测序结果
3 讨 论
HLH是一组由于免疫异常导致的过度炎性反应综合征,本病起病急、进展快,若不能得到及时的诊治可很快死亡[1]。根据病因及发病机制,HLH分为原发性HLH及继发性HLH,前者是由于各种基因缺陷导致的遗传性疾病,也称为家族性HLH,一般认为儿童期多见[5],后者则是在感染、肿瘤或风湿性疾病等基础疾病基础上,启动免疫系统异常活化所导致的反应性疾病[2]。本课题组既往资料显示,217例HLH患儿仅6例为淋巴瘤所致(2.76%),儿童期LAH比例明显低于成人[2,4]。但随着对HLH尤其是LAH的重视,LAH比例似有逐渐增加趋势。
本组患儿均以HLH为首发表现,实验室检查也符合HLH诊断标准[5];患儿均最终确诊为NHL或LPD,故LAH诊断明确。6例患儿中5例发病初期即有淋巴结肿大,但3例系腹腔淋巴结大且病情较重,未能及时进行淋巴结活检确诊。本组患儿骨髓细胞学、活检及FCM均未发现肿瘤细胞,低于文献报道的检出率[4,11],可能与本组患儿样本量较少有关。本组患儿sCD25水平均显著增加,sCD25对诊断LAH并无特异性;3例(50%)LDH显著上升,因此,对淋巴结肿大尤其是伴LDH显著增加的HLH患儿,若病情允许,应积极进行病理检查,尽早明确或排除恶性肿瘤。文献报道成人期LAH通常继发于T或NK细胞来源肿瘤[2,4],本研究中B细胞及T细胞来源肿瘤各50%,可能与样本量较少有关。但儿童与成人LAH基础疾病是否存在差异,尚需要大样本、多中心和较长时间随访才能得出准确的结论。
本研究的病例多数在血液、脑脊液或病理组织中检测到EBV,提示EBV可能在淋巴细胞增殖及其介导的HLH发病及进展中起到重要的作用[12]。本研究中的患儿5例行WES测序,4例存在不同肿瘤致病基因突变,2例存在肿瘤遗传易感基因,但病例数量太少,其临床意义尚需进一步研究证实。
若不能得到及时治疗,HLH患儿可短期内死亡。因此,只要符合HLH标准,即使怀疑LAH,病理活检后也应尽早开始针对HLH的治疗。确诊恶性肿瘤再进行肿瘤相关治疗,可减少疾病早期病死率[1,5]。成人期LAH多推荐化疗CR后进行造血干细胞移植[1],但本研究中的NHL患儿化疗后均可生存,可能因为儿童与成人NHL发病机制及化疗方案选择存在一定差异。LPD所致的LAH目前尚无标准治疗方案,本组3例LPD短期内全部死亡,提示LPD治疗难度高,可能需要个体化的治疗方案,一旦病情好转,尽早行造血干细胞移植可能会提高患者生存率[2,13]。
总之,本研究显示,儿童期由于NHL及LPD等引起HLH并不少见,临床上对于HLH在按照HLH-04方案治疗的同时,需积极寻找病因。LAH患儿发病时往往由于病情过重和(或)缺乏肿大淋巴结,难以早期活检确诊。本研究中的5例患儿进行了WES测序,4例检出肿瘤相关基因突变,理论上NHL或LPD患儿外周血存在循环肿瘤细胞及循环肿瘤DNA[14](ctDNA),如果对于LAH患儿进行外周血ctDNA检查,可能会早期确诊基础疾病和提高疗效。NHL所导致的HLH预后较好,而LPD则预后很差,需要进一步研究寻找更合适的治疗方案。
