耐多药结核病治疗药物及潜在药物的结构和性质计算
2021-06-20李震岳翁约约黄罗仪王朝杰
李震岳,翁约约,,黄罗仪,王朝杰
1.温州医科大学定理临床学院 温州市中心医院 药剂科,浙江 温州 325000;2.温州医科大学 药学院,浙江 温州 325035
2020年WHO发布报告[1]称,2019年全球新发结核病患者约996万,新增150万耐药性结核病患者,包括单药耐药结核病(monodrug-resistant TB,MR-TB)、多耐药结核病(polydrug-resistant TB,PR-TB)、耐多药结核病(multidrug-resistant TB,MDR-TB)和广泛耐药结核病(extensively drugresistant TB,XDR-TB)等[2]。临床常用抗结核药物包括异烟肼、利福平、吡嗪酰胺、乙胺丁醇、链霉素等,备选药物有左氧氟沙星、莫西沙星和阿米卡星等[3],但MDR-TB的全球平均治愈率仅维持在55%左右[4]。
随着耐药结核病患者的增多,研究和开发新的抗结核药物已成为全球消除结核病的重要战略组成部分。ZHAO等[5]报道了基于噁唑烷酮结构合成的17类86种化合物,通过系列体内和体外实验发现,构象限制性的19c比目前临床用于治疗MDR-TB药物利奈唑胺(Linezolid,Lin)[6],正在进行临床试验的舒特唑胺(Sutezolid,Sut)[7]、德帕唑胺(Delpazolid,Del)[8]和TBI-223(223)[9]安全性和疗效更佳。本研究将对上述五种化合物的结构和性质进行计算分析,探索内在关系,促进MDR-TB新药研发。五种化合物的2D结构式见图1。
图1 五种化合物的2D结构式
1 计算方法
药物分子构象采用MMFF94力场进行分子力学计算,在GaussView6.0中的附加模块GMMX中实现。使用默认参数集,总能隙为3.5 kcal·mol-1。当超过10 000次试验的限制,或经过50次连续试验,仍没有新的构象位于能量间隙内全局最低限度,则停止搜索。
理论计算采用密度泛函理论的M06-2X方法,在6-311+G(2d,p)基组水平上对图1五种化合物在真空环境几何结构和电子结构进行优化,再进行红外(IR)振动光谱、紫外-可见(UV-Vis)吸收光谱、电子圆二色(ECD)谱计算。已有文献[10]报道显示,M06-2X这种双杂化元广义梯度近似方法(DH m-GGA)在预测分子几何形状、拉曼强度、热化学和动力学、非共价相互作用等方面表现良好。在上述优化结构的基础上,结合自洽反应场方法中的极化连续介质模型模拟环己烷(cyclohexane,Cyc)、水(water,Wat)和甲醇(methanol,Met)溶剂环境,再用同一基组水平的含时密度泛函理论进行UV-Vis吸收光谱和ECD谱计算,所得波函数用于电子结构分析和概念密度泛函理论系列指数分析,包括分子的亲电/亲核性预测,整体反应性描述符包括电离势(IP)、电子亲和势(EA)、电负性(χ)、整体硬度(η)、化学势(μ)、整体亲电指数(ω)和整体柔软度(S)等计算参照文献[11-13]中定义公式。药代动力学计算借助ACD/Labs Percepta平台实现,该平台基于QSAR(quantitative structure-activity relationship)模型预测化合物的药代动力学部分参数和理化性质。所有量化计算和分析使用Guassian 16程序和Multiwfn 3.8[14]软件完成。
2 结果和讨论
2.1 构象分析 由图1可见,在19c分子中仅有4根键可以旋转产生构象,加上环构象变化,而Sut有5根键可以旋转,Del中4根键,233中6根键,Lin中5根键,各自环的构象变化有差异。运用MMFF94力场搜索显示,在相同的能量范围内,19c仅有21种构象异构体,Sut有48种,Del有63种,223有69种,Lin有56种,可见19c在3aS位实现关环,从而大大减少药效构象,在文献[5]的系列实验中也体现其构象限制带来安全性和疗效的提升。
2.2 几何结构 Lin为噁唑烷酮类抗革兰氏阳性菌药物,同时对MDR-TB具有良好的抗菌活性,本研究的其他四种最典型化合物均以其为基础发展而来。图2绘制了采用M06-2X/6-311+G(2d,p)计算的该五种化合物在真空、环己烷和水环境中的几何结构,主要键长依次标注在相应位置。五种化合物均有3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]oxazin-1-one的立体化学结构核,是抗菌活性的中心。ZHAO等[5]发现(3S,3aS)构型是抗结核活性的最佳构型,即(3S,3aS)-3a,4-dihydro-1H,3H-benzo[b]oxazolo[3,4-d][1,4]-oxazin-1-one核,关键是苯基环上的R1基团和噁唑烷酮环上的R2基团的变化。在R1位引入吗啉环等基团,在R2位取代为氨基、杂环等先后获得86种化合物,19c(见图2)被确定为治疗MDR-/XDR-TB的一种很有潜力的分子。
三种环境计算19c结构中,C=O随着极性增加而逐渐伸长,增加约0.01Å,其余键长变化甚微,饱和硫吗啉环键长变化仅有~0.003Å。其他结构中C=O键长变化与19c类似。Sut是Lin的硫代类似物,其中吗啉基氧被硫取代。MICHALSKA等[15-16]对Sut在环糊精改性非水毛细管电泳的对映选择性识别研究中,采用B3LYP/6-31G*进行了中性态和质子化态的计算,并通过IR光谱、核磁共振和分子模拟解释复合物形成机制。Del是一种第二代噁唑烷酮衍生物,尚在临床试验中。223作为另一种噁唑烷酮类药物正在进行抗结核病的一期临床试验,有关Del和223的计算报道甚少,但结构显示彼此在抗菌核心结构参数较一致。
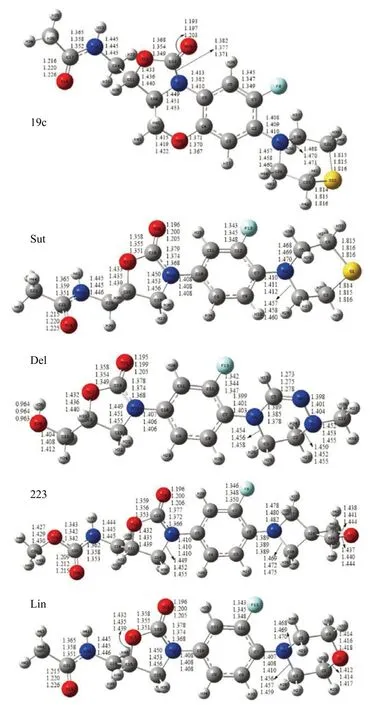
图2 五种化合物在真空、环己烷和水环境中M06-2X/6-311+G(2d,p)优化的主要键长值[单位(Å)]
Lin作为第一个噁唑烷酮抗菌药物,得到广泛研究。实验发现Lin固态物质可能以不同的构象形式存在,根据测定IR和熔点温度,至少有四种构象。对比晶体构型[17]二面角参数,Lin呈第IV种构象中的a分子状时,其ΨN3-C10、ΨC7-N2、ΨC4-C3、ΨC3-N1和ΨN1-C2值分别为-167(1)°、 -172(1)°、 -63(1)°、 -104(1)°和171(2)°,我们用M06-2X/6-311+G(2d,p)在真空计算的数值分别为-165°、-175°、-60°、-76°和172°,彼此吻合较好。a和b分子形态差异在于吗啉环的构象具有互补性。通过计算数据分析,五种化合物核心部分整体受两端基团变化影响不大。溶剂环境对结构影响也不明显。
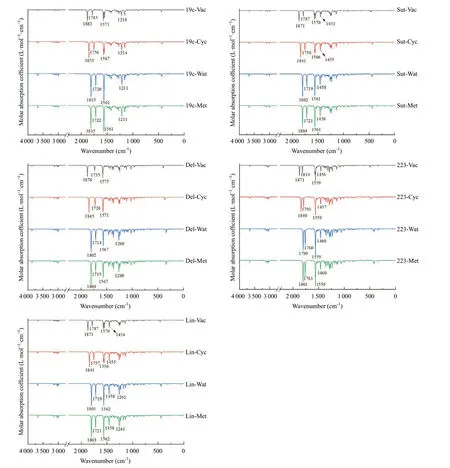
图3 五种化合物在真空、环己烷、水和甲醇环境中M06-2X/6-311+G(2d,p)计算的IR吸收光谱
2.3 电子结构 五种化合物的前线轨道HOMO和LUMO分布分析显示,除Del的HOMO是由三嗪环中氮2p轨道构成的π键,其余都由氟代苯环的π键构成。五种化合物LUMO轨道构成类似,其中19c和Del构成比例和相位一致,Sut、223和Lin构成类似,但占比逐渐降低,在等密度值为0.05情况下,Lin的轨道组成更加分散。五种化合物都有极性,M06-2X/6-311+G(2d,p)计算真空中极性大小是Lin(2.85μ)<Sut(3.00μ)<19c(3.45μ)<223(3.77μ)<Del 6.28μ),水溶剂环境中Lin(4.63μ)<Sut(4.75μ)<19c(5.17μ)<223(5.48μ)<Del(9.18μ),反映出化合物极性越大,在极性环境中极性改变也越大。
2.4 IR光谱 五种化合物中,Lin已应用于临床,针对Lin的各项研究较多[18-19]。我们用M06-2X/6-311+G(2d,p)在四种环境下分别计算了五种化合物的IR振动吸收光谱(见图3),整体而言,在真空、环己烷、甲醇和水环境中,五种化合物对应振动频率基本呈现红移现象,如噁唑烷酮中C=O的频率下降约70 cm-1,酰基中的C=O则下降约55 cm-1,N-H摆动频率受环境影响可忽略。苯环的特征峰整体红外强度比较低。
真空中Lin的两个羰基理论伸缩振动频率为1 871 cm-1和1 787 cm-1,而文献[19]测定VCD数据为1 744 cm-1和1 663 cm-1,两者之间相较,理论计算的校正因子为0.930。MICHALSKA等[15]采用IR和NMR手段分析了Sut在环糊精β-CD中手性识别的谱学特征,并用B3LYP/6-31G*方法计算了单一Sut分子和用HF/6-31G*计算了复合体系。Sut的特征峰在噁唑烷酮的C-O键1 012 cm-1,噁唑烷酮的CH2对称摆动1 416 cm-1,乙酰氨基中的C-N伸缩振动频率1 518 cm-1,而C=O键在1 665 cm-1和1 745 cm-1出现。对比我们计算的Sut在甲醇中IR吸收谱,C=O键在1 721 cm-1和1 804 cm-1出现,而特征峰分别在1 045 cm-1、1 457 cm-1和1 560 cm-1,彼此吻合非常好。此后MICHALSKA等[16]报道了Sut合成中间体的IR和Raman数值,结合B3LYP/6-311G(d,p)计算分析,对比本研究M06-2X/6-311+G(2d,p)计算数值,基本上高5~40 cm-1。
2.5 UV-Vis光谱 五种化合物用M06-2X/6-311+G(2d,p)在真空、环己烷、水和甲醇中计算得到的UV-Vis吸收光谱曲线见图4。五种化合物在150~320 nm范围均有两个吸收峰,其中远紫外区180~200 nm和近紫外区220~320 nm理论计算各有一个吸收峰。溶剂效应对223影响最小,对Sut和Del吸收峰强度影响最为明显,呈现远紫外蓝移近紫外红移大约5 nm,这有待实验验证。紫外吸收光谱来源主要是HOMO轨道电子向LUMO+2跃迁,而Del则是HOMO轨道电子向LUMO跃迁,贡献率达到86%。
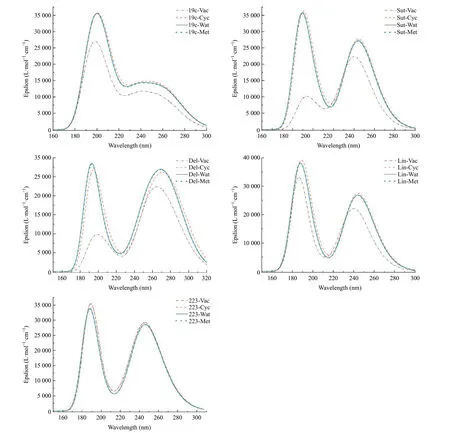
图4 五种化合物在真空、环己烷、水和甲醛环境中M06-2X/6-311+G(2d,p)计算的UV-Vis吸收光谱
在几何结构分析部分,显示Lin是IV型中的a分子态,在甲醇中近紫外最大吸收峰对应的是242.3 nm,与实验[18]上用245.6 nm测定值接近。MICHALSKA等[16]实验显示Sut的R和S构型在水中时在200 nm和250 nm处有吸收峰,在甲醇中时,R和S构型可能在190 nm和260 nm处有最大吸收峰,另外溶剂环境对UV-Vis吸收光谱影响差异明显,而我们计算溶剂模型简单,区分不明显,但计算的最大吸收波数比较接近。其他化合物的实验报道甚少,化合物19c UV-Vis吸收光谱实验数据未见报道。
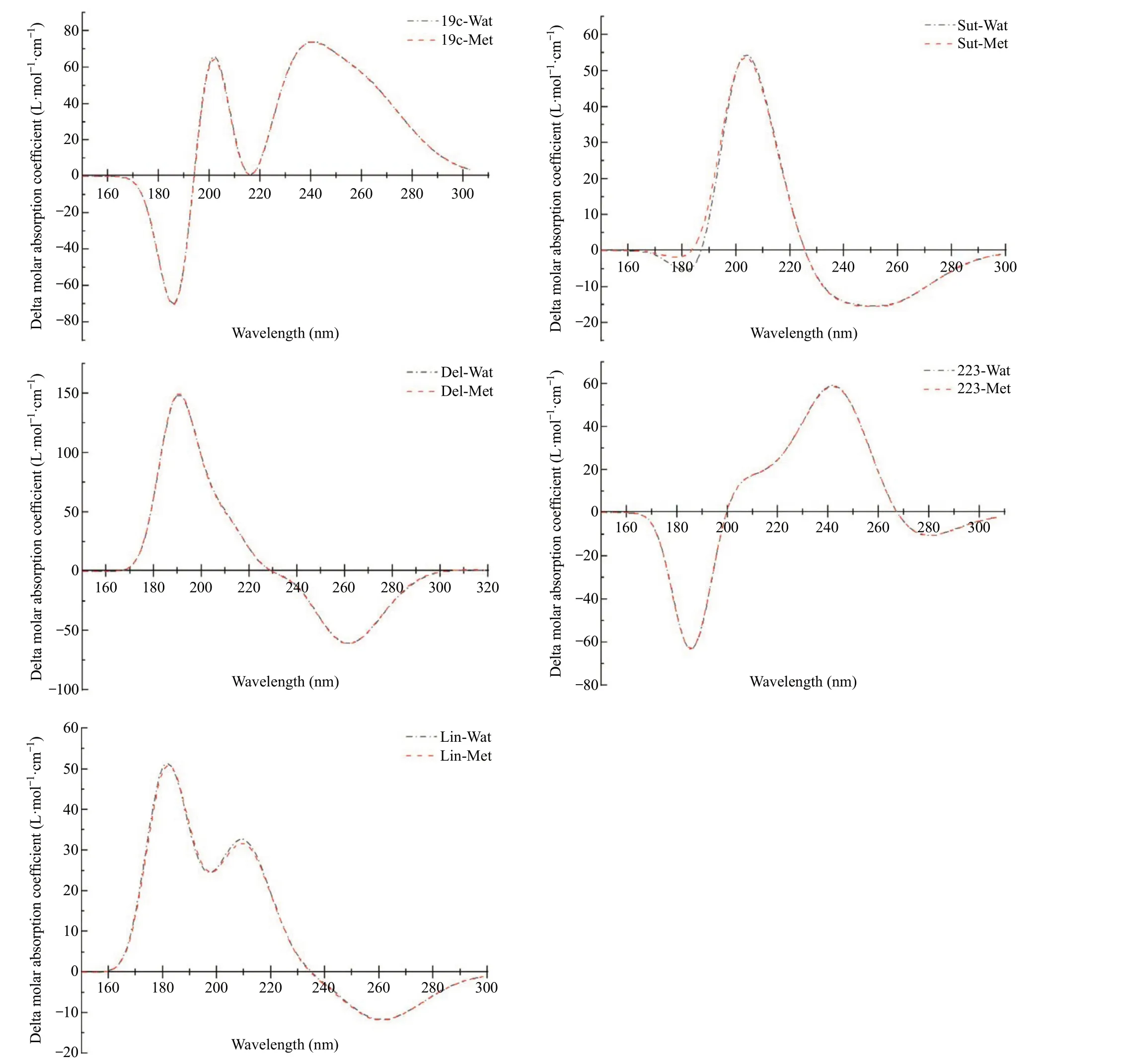
图5 五种化合物在水和甲醇中M06-2X/6-311+G(2d,p)计算的ECD谱
2.6 ECD分析 由于五种化合物中都至少有一个手性中心,而19c是两个,分别是3S和3aS。目前计算显示五种化合物在水中和甲醇中的ECD谱是高度吻合,见图5。由于其整体结构差异明显,五种化合物的ECD峰形不同,而手性微环境Sut、223和Lin比较一致。MICHALSKA等[15]在手性识别实验中测定Sut在水和甲醇中的ECD,可能存在构型变化,结果认为图谱不适合其实验解释,后来他们对Sut手性池合成中间体和终产物进行谱学分析[16]。我们用M06-2X/6-311+G(2d,p)方法结合极化连续介质模型计算甲醇中的单一构象的ECD,在CES分布上与实验一致,波数上也十分接近。FRELEK等[19]对Lin进行ECD和VCD测定,主要是构象II和III,而我们计算比较确认是构象IV中a分子态。对ECD曲线的细致分析表明,在构象II中,在221 nm左右存在一个相对较强的负带,并且在构象III的ECD光谱中没有这种负带。因此,这个带可以被认为是区分Lin的两种构象的诊断带。其他三种化合物实验测定报道较少,我们的计算有待实验测定。
2.7 分子静电势分布图 五种抗MDR-TB分子表面静电势如图6所示,蓝色区域表示负电荷区域,易受到亲电试剂的进攻;红色区域表示正电荷区域,易受到亲核试剂的进攻;白色表示零电位区域。其中,黄球对应静电势极大值点,青球对应极小值点。根据图6,我们发现静电势的极小值点大多位于噁唑烷酮的羰基氧、乙酰基氧和三嗪环氮附近,而极大值点大多位于氨基氢、亚甲基和端甲基氢附近。同时发现19c、Sut与Lin静电势分布比较接近,而Del出现6处极值点,223有5个位点。
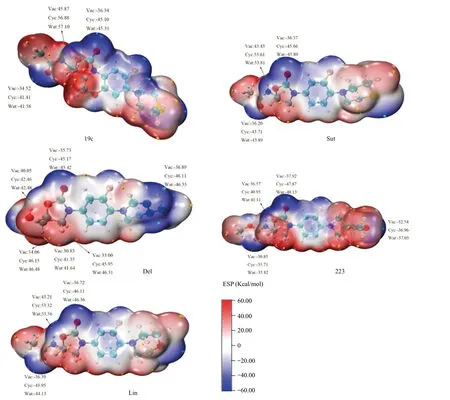
图6 五种化合物在真空、环己烷和水环境中M06-2X/6-311+G(2d,p)计算的静电势图
2.8 全局反应性描述符 五种化合物的全局反应性参数见表1。以Lin为参考,真空中Sut的μ最低,说明其稳定性较好,19c居中;在水环境中则是19c最低。在水环境中19c、Sut和Lin的化合物整体硬度η较大且比较接近,说明它们对微干扰下化学体系电子云畸变的抵抗能力较强,分子稳定性较高;整体亲电指数随着环境极性增加而下降,化合物19c在水中ω表明其得电子能力强,反应活性较高。前线轨道能差19c与Lin比较接近,EA值均比较小或为负值,说明五种化合物整体对电子的束缚能力较弱,得电子的趋势很小。从全局反应性描述符分析,五种分子间差距都较小,作为同类药物具有很高的相似性,也客观反映出Lin作为已经使用的药物的合理性,其余在进行临床研究的化合物,具有很大的成药潜力。
2.9 药代动力学评价 基础实验显示具有潜在药效的许多化合物因安全性、有效性和药代动力学等问题而被淘汰。通过ACD/Labs Percepta软件,可预测化合物的吸收、分布、代谢、排泄、毒性及理化性质等方面性质。如Lipinski规则是广泛用于判定化合物类药性是否良好的经验规则,类药性的筛选有利于获得药动学、药效学性质平衡的候选药。计算发现五种化合物均符合Lipinski规则,类药性较好,化合物19c在胃(logD-pH 1.70)和血液(logD-pH 7.40)中分布系数比Lin更优。
药代动力学性质是成药的重要指标之一[20],五种化合物的药动学相关参数预测结果见表2。五种化合物均不是糖蛋白(P-gp)的底物(P-gp是一种外排转运蛋白,表达于各种具有分泌功能的细胞上,如肠上皮细胞和脑毛细血管内皮细胞上[21])。五种化合物对肝脏的主要代谢酶(CYP1A2、CYP2C9、CYP2C19、CYP2D6和CYP3A4)的抑制作用都比较弱或无抑制作用,Lin对五种代谢酶整体抑制作用最弱,19c对CYP1A2亚型,化合物19c和Sut对CYP3A4亚型稍微有抑制作用。基因毒性(Ames反应)指标显示五种化合物可能有轻微的基因毒性。hERG指标反映五种化合物可能有轻微的心脏毒性。Caco-2(Caco-2细胞内有药物代谢酶,Caco-2细胞模型可用于研究药物转运机制和预测药物肠吸收的体外筛选[22])结果表明Del化合物的吸收性极差,其余均相近,19c优于Lin。血浆蛋白结合率(plasma protein binding,PPB)会影响半衰期、清除率、药物分布等药代动力学参数[23]。19c和Sut与血浆蛋白结合强度中等,而Lin与血浆蛋白结合能力较弱。五种化合物在代谢稳定性方面比较接近。而ZHAO等[5]的系列细胞和动物实验,19c与Sut和Lin相比均接近或更优,而体外抗结核菌最低抑菌浓度(minimum inhibitory concentration,MIC)更低,体外抗耐药结核菌比Sut和Lin的MIC要低2~10倍,hERG K+毒性在30 mmol/L以上。一项为期4周的大鼠毒理学研究显示,19c比Lin骨髓抑制作用更小,为噁唑烷酮类的突破。化合物19c在小鼠结核病菌感染模型中体内疗效优于Lin和Sut。五种化合物穿过血脑屏障都不易,在中枢神经系统(central nervous system,CNS)中的浓度低,Del在肠道吸收(human intestinal absorption,HIA)比其他四种弱。
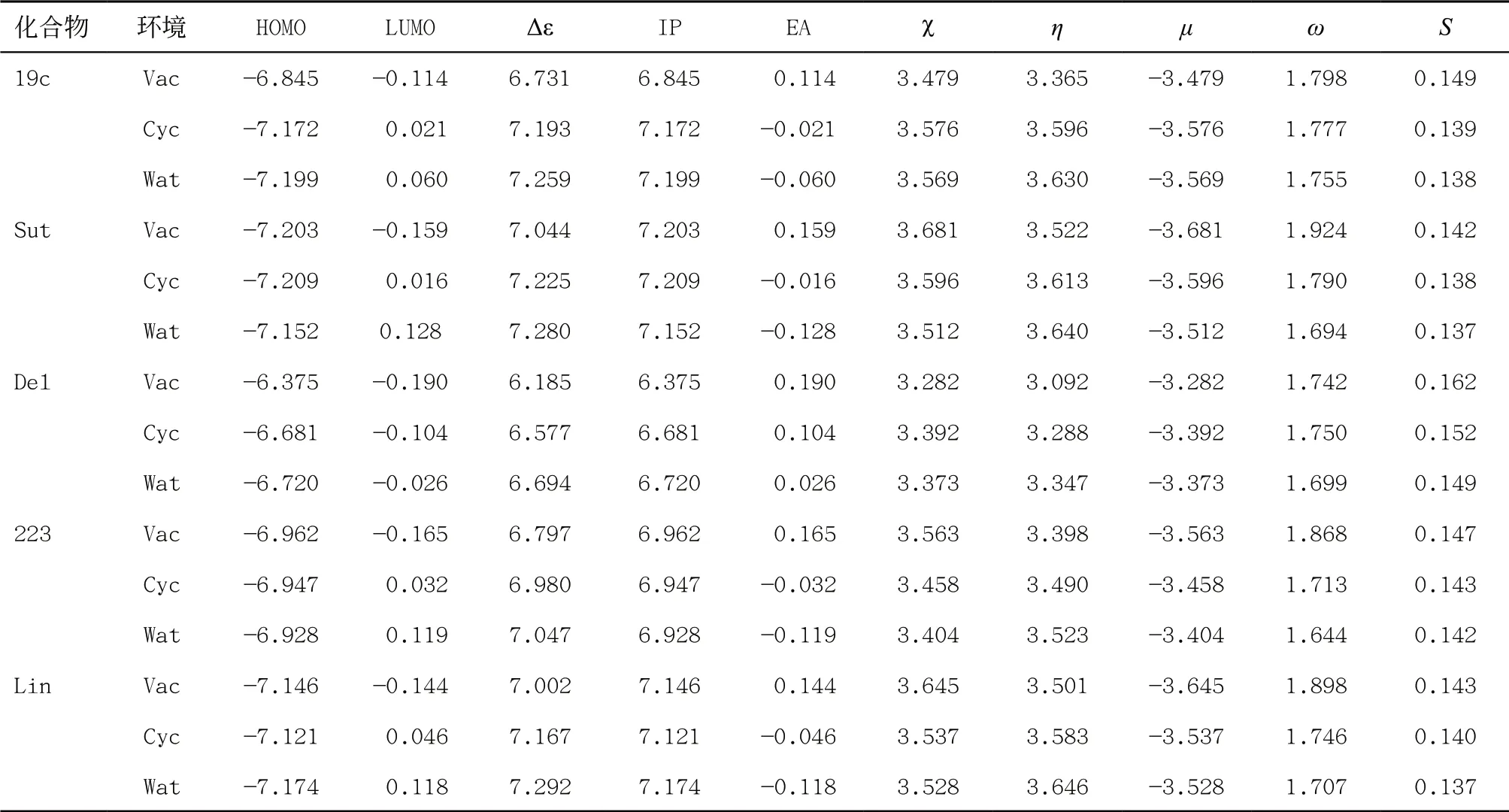
表1 五种化合物在真空、环己烷和水环境中M06-2X/6-311+G(2d,p)计算的全局反应性描述符(eV)
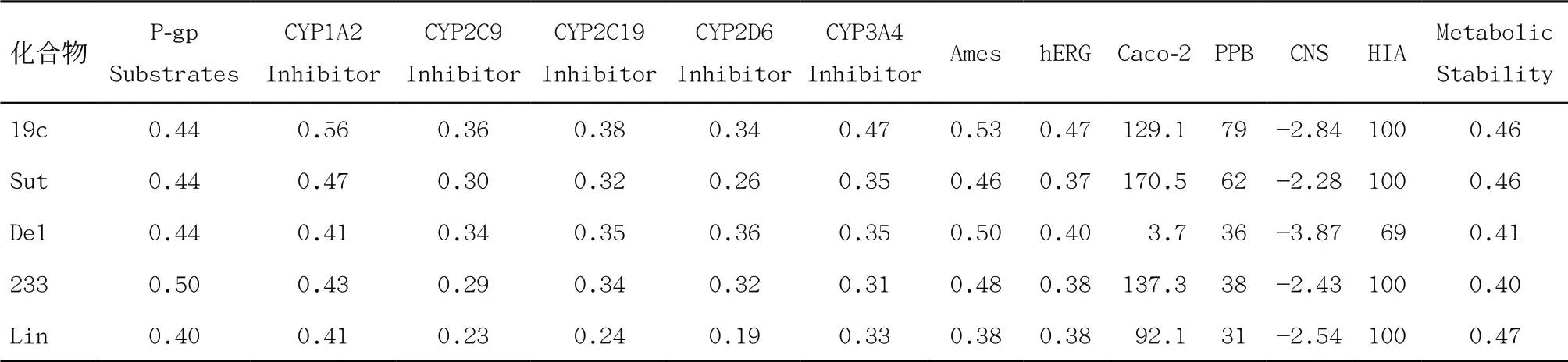
表2 五种化合物的药代动力学参数
综上所述,本研究在M06-2X/6-311+G(2d,p)水平下,对五种MDR-TB治疗药物和潜在药物化合物的药效构象、几何和电子结构、IR等系列谱学性质进行了计算分析,并结合概念密度泛函数预测整体反应指数,再利用ACD/Labs Percepta软件预测化合物的ADME/Tox和成药性。新化合物19c增加一个手性中心形成环状结构大大减少药效构象,五种化合物都是噁唑烷酮类抗结核菌化合物,在不同溶剂环境中,药效结构几何参数值基本一致,与晶体参数吻合较好。极性环境使Del极性改变最大,其HOMO分布与其他四种不同。该方法计算的IR光谱采用0.930校正因子后与实验和其他计算方法吻合很好。Lin的计算最大吸收波数与实验完全一致,除Del紫外吸收光谱是HOMO电子向LUMO跃迁为主外,其他均是HOMO向LUMO+2跃迁为主,都具有双峰曲线。Sut计算的ECD峰与实验相吻合。19c、Sut和Lin静电势分布主要集中在噁唑烷酮端,而Del和223另一端呈电势负性。8种全局反应性描述符显示五种化合物彼此数值接近,具有相同母核的不同衍生物,具有明显的类药性。类药性评价显示Del分布系数与其他差别大外,基本比较接近。药代动力学参数五种化合物比较一致,但临床用药Lin的参数更优。
