一测多评法同时测定蒙药扎冲十三味丸中8种成分
2021-06-11宝鲁尔吴杰斯白塔那陈红梅王秀兰
宝鲁尔,王 徽,吴杰斯,白塔那,陈红梅,王秀兰*
1.内蒙古民族大学蒙医药学院,内蒙古 通辽 028000
2.内蒙古民族大学附属医院,内蒙古 通辽 028000
扎冲十三味丸(Zhachong Shisanwei Pills,ZSP)又名嘎日迪十三味丸、十三味麝香丸、十三味大鹏金翅丸、扎冲朱素木,在蒙医临床上使用历史较长,此方由木香、诃子、甘草、麝香、肉豆蔻、制草乌、石菖蒲、丁香、沉香、珍珠(制)、磁石(煅)、禹粮土、珊瑚(制)共13 味单药组成,具有除“协日乌素”、镇静安神、祛风通窍、舒筋活血功效,用于左痰右痪、手脚麻痹、腰腿不利、言语不清、口眼歪斜、神经麻痹、半身不遂、筋骨疼痛、关节疼痛、风湿[1]。临床上用于治疗缺血性脑血管病(白脉病)、风湿性关节炎(协日乌素病)、面肌痉挛、周围性神经麻痹、坐骨神经痛、冠心病心绞痛等疾病[2-4]。
蒙药是蒙古族历代蒙医经典医籍传承的宝贵财富,具有独特的功效。近几年研究蒙药复方的学者和研究人员逐渐增多,同时,为了顺应国际和国内的用药需求,对蒙药复方的质量控制也不再是单一成分的含量测定,而通过对多成分同时测定实现蒙药复方的多指标质量评价。一测多评法(quantitative analysis of multi-components by single-marker,QAMS)是一种采用1 个成分来实现对多个成分同步监控的有效方法,近年来在蒙药复方制剂多指标成分定量测定中得到广泛应用。本实验采用QAMS法同时测定ZSP中主要活性成分没食子酸、鞣花酸、绿原酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵、沉香四醇的含量,为蒙药ZSP 的多指标质量控制提供实验依据和参考方法。
1 仪器与材料
1.1 仪器
Agilent 1260 Infinity II 型高效液相,美国Agilent科技有限公司;Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5 μm);Inertsil ODS-3 柱(250 mm×4.6 mm,5 μm);BDS HypersilTMC18柱(250 mm×4.6 mm,5 μm);岛津LC-20A(20A)型高效液相,日本岛津科技有限公司;QUINTIX35-1CN 型十万分之一电子天平,赛多利斯科学仪器(北京)有限公司;KQ-300VDY 型医用超声波清洗器,昆山市超声仪器有限公司。
1.2 材料
ZSP,9 个批次,批号1908003、1908005、1908008、1909010、1909011、1909016、1911004、1911005、1911006,内蒙古奥特奇蒙药股份有限公司。对照品没食子酸(批号110831-201605,质量分数90.8%)、绿原酸(批号110753-200413,质量分数99.3%)、鞣花酸(批号111959-201903,质量分数88.8%)、沉香四醇(批号111980-201904,质量分数98.6%)、丁香酚(批号110725-201917,质量分数99.1%)、甘草酸铵(批号110731-201619,质量分数93.0%)、α-细辛脑(批号100298-201203,质量分数100.0%)、β-细辛醚(批号112018-201802,质量分数99.3%)均购于中国食品药品检定研究院。甲醇为色谱纯;水为超纯水;其他试剂均为分析纯。
2 方法与结果
2.1 HPLC 定量方法
2.1.1 色谱条件 Inertsil ODS-3 柱(250 mm×4.6 mm,5 μm),流动相为甲醇-0.1%磷酸水溶液,梯度洗脱:0~7 min,30%甲醇;7~20 min,30%~51%甲醇;20~28 min,51%~59%甲醇;28~55 min,59%~75%甲醇;检测波长为0~7 min,271 nm(没食子酸);7~20 min,252 nm(绿原酸、沉香四醇);20~28 min,254 nm(鞣花酸);28~33 min,281 nm(丁香酚);33~45 min,257 nm(β-细辛醚、α-细辛脑);45~55 min,237 nm(甘草酸铵);体积流量1.0 mL/min;柱温30 ℃;进样量7 μL。该色谱条件下对照品及样品色谱图见图1。
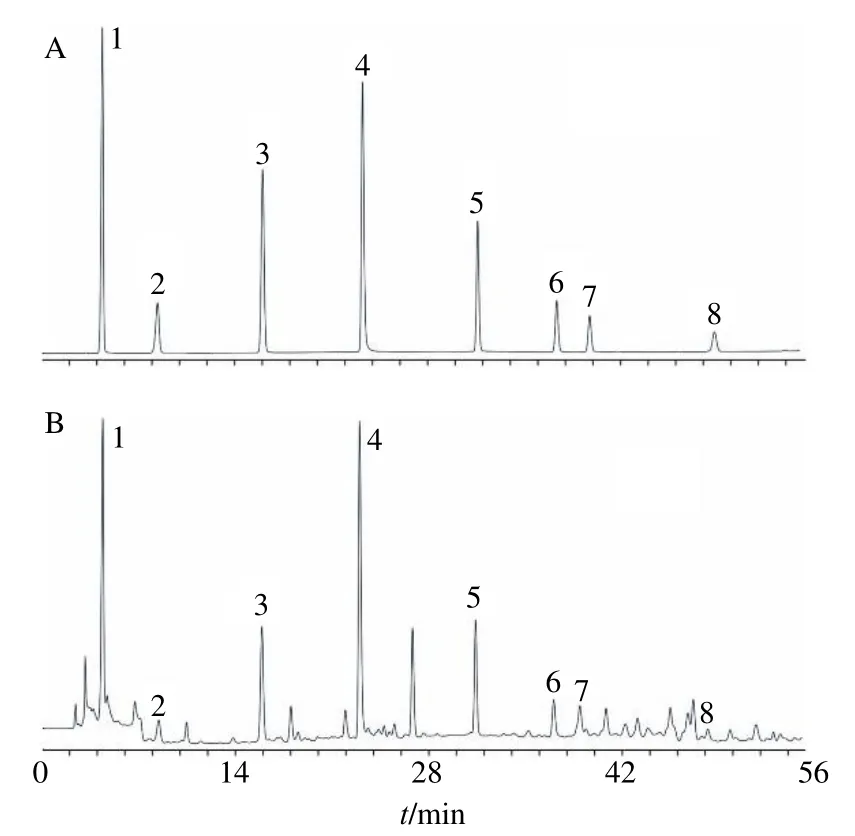
图1 混合对照品 (A) 和ZSP 样品 (B) 的HPLC 图Fig.1 HPLC of mixed reference substances (A) and ZSP sample (B)
2.1.2 混合对照品溶液制备 分别精密量取没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵对照品储备液适量,混合均匀,加甲醇进行稀释,制成含没食子酸0.184 mg/mL、绿原酸0.116 mg/mL、沉香四醇0.167 mg/mL、鞣花酸0.063 mg/mL、丁香酚0.207 mg/mL、β-细辛醚0.042 mg/mL、α-细辛脑0.025 mg/mL、甘草酸铵0.094 mg/mL 的混合对照品溶液。
2.1.3 供试品溶液制备 精密称取样品2.0 g,置于具塞锥形瓶中,精密加入甲醇25 mL,密塞,称定 质量,超声35 min,放冷至室温,甲醇补足减失的质量,摇匀,滤过,取续滤液经0.45 µm 微孔滤膜滤过,即得供试品溶液。
2.1.4 线性关系考察 将“2.1.2”项下混合对照品溶液按照“2.1.1”项下色谱条件分别进样0.5、2.0、3.5、5.0、6.5、8.0 μL,记录色谱峰面积。以峰面积为纵坐标(Y),对照品质量为横坐标(X)进行线性回归,各组分的回归方程及相关系数见表1。可见该方法在试验范围内线性关系良好。
2.1.5 精密度试验 精密吸取“2.1.2”项下混合对照品溶液,以“2.1.1”项下色谱条件重复进样6 次,记录峰面积,没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵峰面积的RSD 值分别为0.40%、0.73%、0.35%、0.51%、0.93%、0.79%、1.81%、1.69%,表明在该条件下仪器精密度良好。
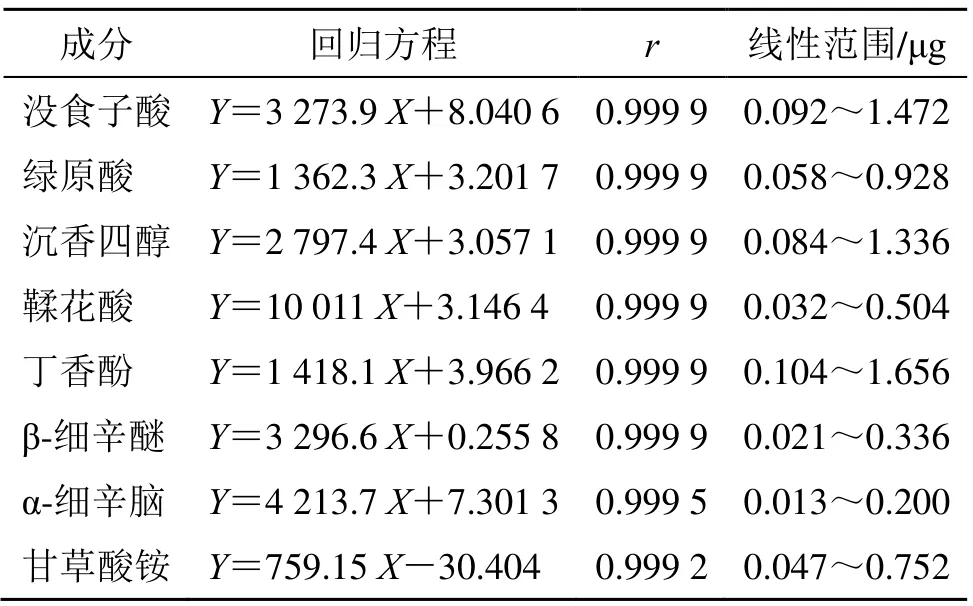
表1 线性关系测定结果Table 1 Results of determination of linearity relation
2.1.6 稳定性试验 取ZSP 粉末2.0 g,精密称定,按照“2.1.3”项下方法制备供试品溶液,室温放置,分别于0、2、4、6、8、10、12、24 h 按照“2.1.1”项下色谱条件进样,记录峰面积,没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵峰面积的RSD 值分别为0.14%、1.08%、0.22%、0.08%、0.72%、0.47%、1.84%、1.71%,表明该供试品溶液在室温下24 h 内稳定。
2.1.7 重复性试验 取同一批次ZSP 粉末(批号1908003)2.0 g,精密称定,按照“2.1.3”项下方法制备6 份供试品溶液,按照“2.1.1”项下色谱条件进样,计算没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵峰面积的RSD 值分别为0.06%、1.03%、0.19%、0.12%、0.29%、0.57%、1.86%、1.54%,表明该方法重复性良好。
2.1.8 加样回收率试验 取同一批次ZSP 粉末(批号1908003)共6 份,每份1.0 g,精密称定,置50 mL 具塞锥形瓶中,精密量取质量浓度为 1.66 mg/mL 没食子酸、0.52 mg/mL 绿原酸、0.97 mg/mL沉香四醇、0.67 mg/mL 鞣花酸、1.74 mg/mL 丁香酚、0.29 mg/mL β-细辛醚、0.26 mg/mL α-细辛脑、0.56 mg/mL 甘草酸铵的混合对照品溶液1 mL,再按照“2.1.3”项下方法制备供试品溶液,按“2.1.1”项下色谱条件进样,记录峰面积,计算8 种成分的加样回收率以及RSD 值,结果显示,没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵的平均加样回收率分别为 94.49%、100.43%、91.90%、94.66%、99.82%、95.85%、99.66%、97.34%,RSD 值分别为0.45%、0.42%、0.26%、0.64%、0.60%、1.37%、0.78%、1.95%,表明该方法具有良好的回收率。
2.2 相对校正因子(fs/i)的确定及耐用性考察
2.2.1fs/i的测定 取“2.1.2”项下混合对照品溶液进样分析,记录各成分的峰面积,以丁香酚为内参物,取多个质量点计算所得的fs/i,取平均值作为定量用fs/i[fs/i=fs/fi=AsCi/AiCs,其中As为丁香酚对照品的峰面积,Cs为丁香酚对照品的质量浓度,Ai为待测成分的峰面积,Ci为待测成分的质量浓度][5-7]。计算待测组分没食子酸、绿原酸、沉香四醇、鞣花酸、β-细辛醚、α-细辛脑、甘草酸铵的fs/i,结果见表2。结果显示,没食子酸、绿原酸、沉香四醇、鞣花酸、β-细辛醚、α-细辛脑、甘草酸铵与内参物丁香酚之间的fs/i分别为0.433 4、1.068 3、0.505 6、0.141 6、0.429 0、0.346 2、1.748 8,RSD 值分别为0.77%、0.40%、0.90%、0.82%、0.61%、1.42%、1.69%。
2.2.2 不同色谱仪器和色谱柱对fs/i的影响 本实验考察了Agilent 1260 高效液相色谱仪器(美国)和LC-20A(20A)高效液相色谱仪(日本岛津)对fs/i的影响,Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5 μm)、Inertsil ODS-3 柱(250 mm×4.6 mm,5 μm)、BDS HypersilTMC18柱(250 mm×4.6 mm,5 μm)对fs/i的影响,结果见表3。结果显示,2 台色谱仪器和3 根色谱柱测得各待测成分fs/i的RSD均小于2.0%,表明fs/i在不同色谱仪器和色谱柱下具有良好的耐用性。
2.2.3 不同柱温对fs/i的影响 本实验考察了不同柱温(25、30、35、40 ℃)对fs/i的影响,结果见表4。结果显示,在不同柱温测得各待测成分fs/i的RSD 均小于2%,表明fs/i在柱温有波动时具有良好的耐用性。
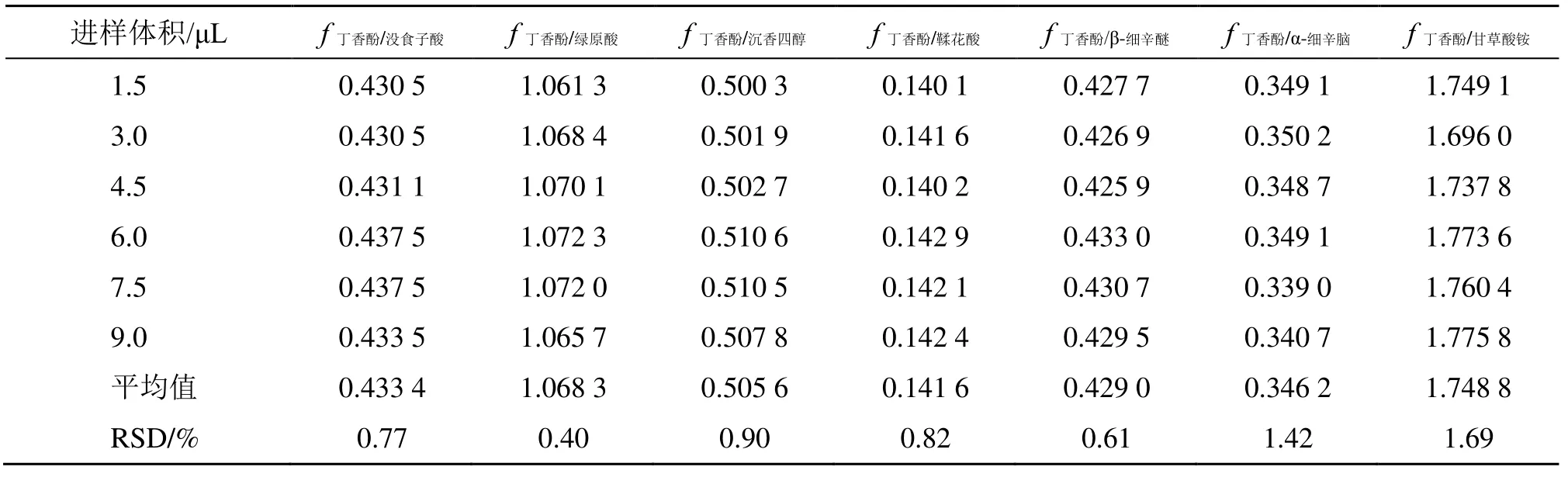
表2 ZSP 中7 种成分的fs/iTable 2 fs/i of seven components for ZSP
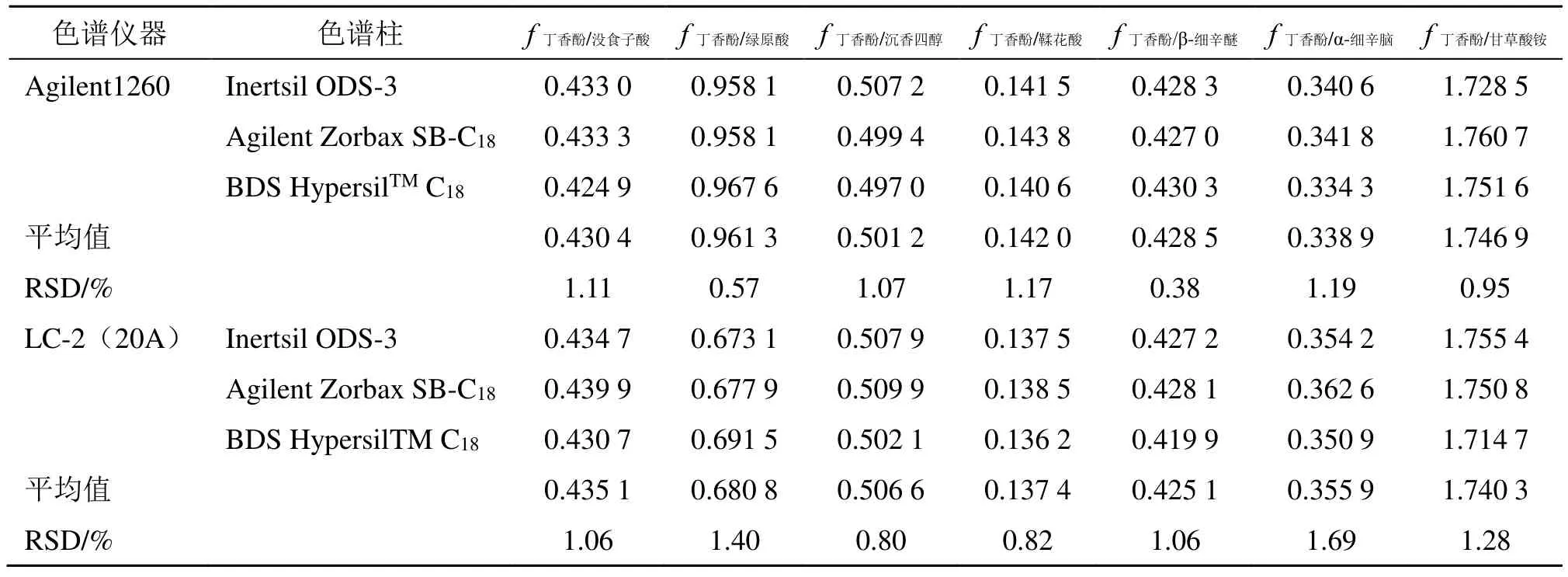
表3 不同色谱仪器和色谱柱对fs/i 的影响Table 3 Effects of different instruments and columns on fs/i
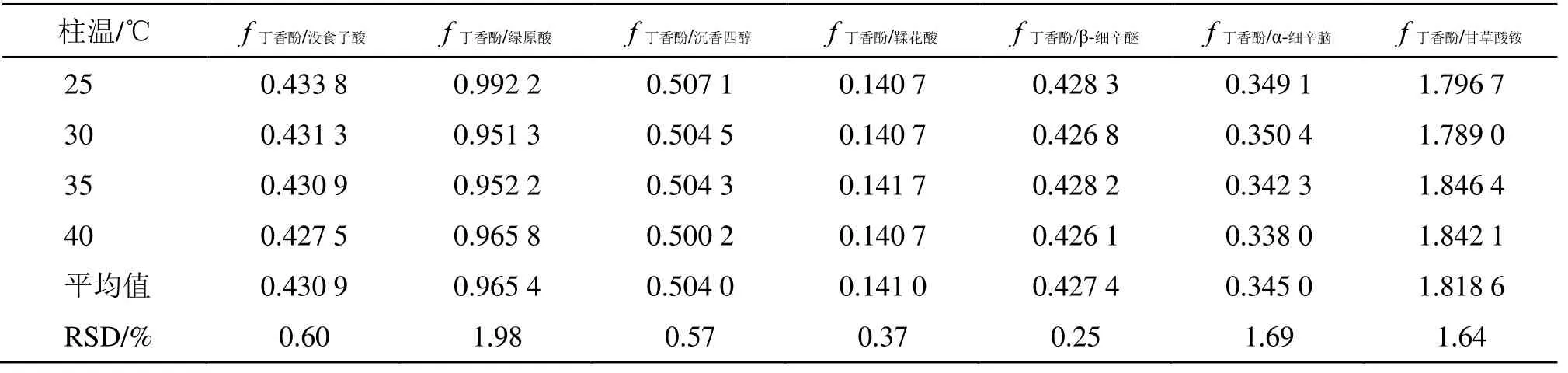
表4 不同柱温对fs/i 的影响Table 4 Effects of different column temperatures on fs/i
2.2.4 不同体积流量对fs/i的影响 本试验考察了不同体积流量(0.8、1.0、1.2、1.4 mL/min)对fs/i的影响,结果见表5。结果显示,在不同体积流量测得各待测成分fs/i的RSD 均小于2%,表明fs/i在体积流量有变化时具有良好的耐用性。
2.3 待测组分色谱峰定位
文献对色谱峰的定位普遍采用相对保留时间和保留时间差来定位峰[8-11]。取混合对照品溶液,选取2 台色谱仪器和3 根不同型号色谱柱,按“2.2.1”项下色谱条件进行测定,以丁香酚为参照峰,计算其他7 种成分的相对保留时间(ti/s)和保留时间差(Δti/s)。结果见表6、7。
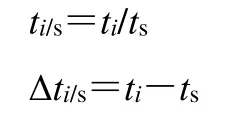
s 为内参物,i为待测组分,t为保留时间
表6 的结果显示,在相同色谱条件下,当色谱柱发生变化时,没食子酸、绿原酸、沉香四醇、鞣花酸相对保留时间的RSD 值均大于5%,表明不能用相对保留时间定位色谱峰。
表7 的结果显示,在相同色谱条件下,没食子酸、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵的保留时间差相差较大,其RSD 值均大于5%,表明不能用保留时间差定位色谱峰。
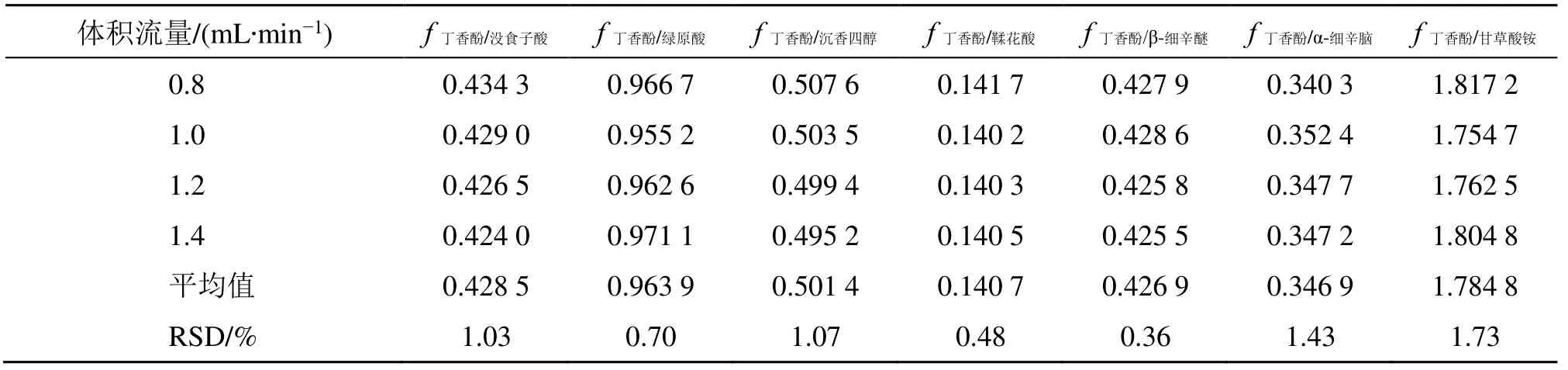
表5 体积流量对fs/i 的影响Table 5 Effects of volume flow rate on fs/i
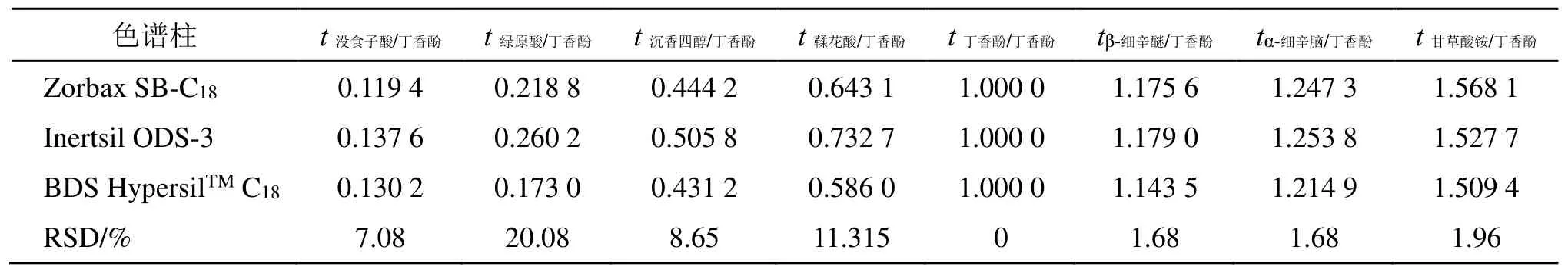
表6 不同色谱柱测得的ti/sTable 6 ti/s of different columns
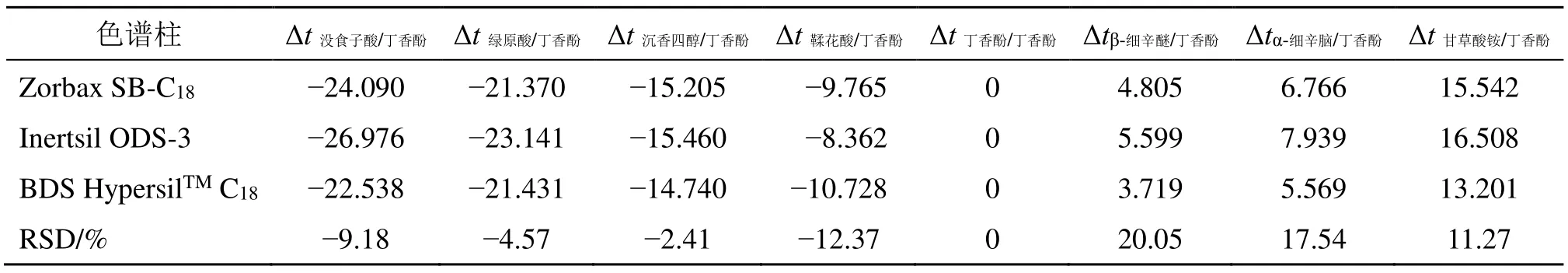
表7 不同色谱柱测得的Δti/sTable 7 Difference of retention time measured by different chromatographic columns
本实验进一步采用两点校正法[12-13]定位色谱峰。精密吸取“2.1.2”项下混合对照品溶液7 μL,以确定的色谱条件分别在3 根色谱柱上测定,分别得到没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵的保留时间,以Agilent 1260 仪器和Inertsil ODS-3 色谱柱为标准仪器和标准色谱柱,以此仪器和色谱柱测定的没食子酸和甘草酸铵的保留时间为横坐标(x),其他仪器和色谱柱测定的没食子酸和甘草酸铵的保留时间为纵坐标(y),分别建立线性方程,再将标准仪器和标准色谱柱上的绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑的保留时间为自变量,分别代入各对应色谱柱线性方程中,求得绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑在相应的各色谱柱上的理论预测保留时间,将计算得到的各待测组分理论预测保留时间与该组分在相应柱上实测的保留时间差为绝对误差(absolute error,AE,AE=测量值-真实值),结果见表8。结果显示,在色谱条件相同,色谱仪器和色谱柱不同时,没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵的实测保留时间与理论预测保留时间的差值较小,故本实验用两点校正法来定位色谱峰。

表8 不同仪器和色谱柱时预测出峰时间Table 8 Peak time with different instruments and different chromatographic columns
2.4 QAMS 法与外标法结果对比
取2.0 g ZSP 样品粉末,精密称定9 份,按照“2.2.3”项下方法制备供试品溶液,在“2.2.1”项下色谱条件进样检测,记录峰面积,采用QAMS 法建立内参物丁香酚与没食子酸、绿原酸、沉香四醇、鞣花酸、β-细辛醚、α-细辛脑、甘草酸铵的相对校正因子,对9 个批号ZSP 样品中各成分含量进行计算,同时采用外标法对没食子酸、绿原酸、沉香四醇、鞣花酸、β-细辛醚、α-细辛脑、甘草酸铵成分进行含量测定,并以相对误差(relative error,RE)为参数对2 种方法所得结果进行比较[14],结果见表9。结果显示,QAMS 法与外标法所测定结果之间差异的RE 均小于5%,表明2 种方法测之间差异较小,所以本实验之前建立的fs/i准确可靠,可以用于测定蒙药ZSP 中没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵的含量。
3 讨论
3.1 蒙药ZSP 测定指标成分的选定
蒙药ZSP含量测定指标及其测定方法文献已有报道,但所选择的测定指标均为一种药材中的一种或者多种指标。本研究建立了多种药材中的多指标成分测定方法,采用高效液相色谱法同时测定了没食子酸、绿原酸、沉香四醇、鞣花酸、丁香酚、β-细辛醚、α-细辛脑、甘草酸铵的含量,用以表征蒙药ZSP 的质量,能更加全面地反映安全质量。
3.2 供试品制备条件的选定
本实验考察了提取溶剂(甲醇、75%甲醇、乙腈),超声时间(25、35、45 min),以所测8 个成分的提取效率为指标,最终确定ZSP 供试品溶液的制备方法为以甲醇为提取溶剂、超声提取时间为35 min,过滤纸过滤方法来处理供试品。
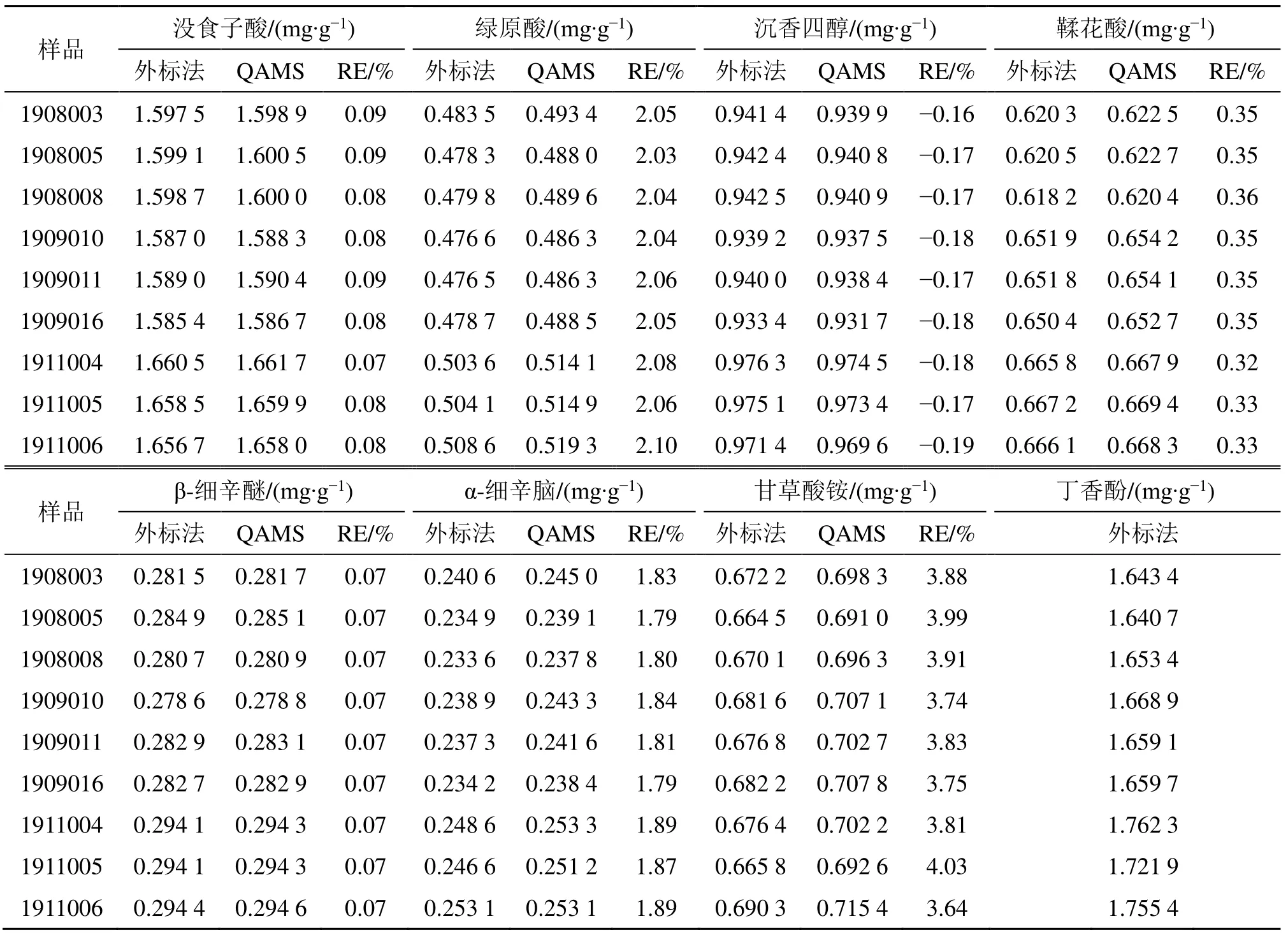
表9 QAMS 法与外标法测得9 批ZSP 中8 种成分的质量分数 (n = 9)Table 9 Content of four constituents in three batches of ZSP by QAMS and external reference method (n = 9)
3.3 色谱条件的选定
在流动相条件摸索中,考察了甲醇-水、甲醇- 0.05%磷酸水溶液、乙腈-0.05%磷酸水溶液、甲醇- 0.1%磷酸水溶液流动相体系,以ZSP 中所测8 种成分的分离效果为指标,最终确定以甲醇为流动相C、以0.1%磷酸水溶液为流动相D,考察不同体积流量(0.8、1.0、1.2、1.4 mL/min)、不同柱温(25、30、35、40 ℃)、不同进样量(5、7、10、15、20 µL),在保证各色谱峰分离度的前提下,尽量缩短分析时间,确定最终条件为甲醇-0.1%磷酸水溶液梯度洗脱,体积流量1.0 mL/min,柱温30℃,进样量7 µL。
3.4 检测波长的选定
蒙药复方制剂所含成分复杂,各成分的最大吸收波长不同,当采用某一个固定波长时,难以确保所测出各成分的灵敏度和精密度、获得最大吸收,影响定量测定结果[15]。本实验采用DAD 检测器在190~400 nm 进行光谱扫描,对不同波长下检测得的三维图谱进行分析对比。结果显示,没食子酸的最大吸收波长为271 nm、绿原酸与沉香四醇的最大吸收波长为252 nm、鞣花酸的最大吸收波长为254 nm、丁香酚的最大吸收波长为281 nm、β-细辛醚与α-细辛脑的最大吸收波长为257 nm,甘草酸铵的最大吸收波长为237 nm。为了使各成分在同一色谱图中均被采集,故采用VWD 检测器切换波长法进行含量测定[16]。
3.5 内参物的选定
建立一测多评法中选定好内参物是很重要,选定内参物的标准是易得、价廉、有效者、峰面积和保留时间稳定、供试品中的含量高,故本实验选定丁香酚为内参物[17]。
3.6 色谱峰定位法的选定
各待测成分色谱峰的准确定位是一测多评法成功应用的关键之一。根据文献报道[18],大多数采用相对保留时间和保留时间差法定位待测成分色谱峰。本实验当中考察了相对保留时间法和保留时间差法、两点校正法在不同厂家仪器和色谱柱中的重现性。实验结果显示,在不同色谱仪和不同色谱柱测得的各待测成分出峰时间有所不同,相对保留时间法和保留时间差法的RSD 均大于5%,故不适合对本实验一测多评法色谱峰定位。采用两点校正法进行定位色谱峰时,所得各待测成分色谱峰实测保留时间与预测保留时间差值较小,最终本实验选定采用两点校正法定位待测成分色谱峰。
本实验建立QAMS 法同时测定ZSP(9 个批次)中8种成分的含量,对QAMS法的可行性进行探讨,结果表明QAMS 法测得结果与外标法法测得结果没有显著差异,表明在对照品难以得到的情况下可以通过测定蒙药ZSP中丁香酚的含量来计算出其他7 种成分的含量,实现多指标同步质量控制,反映蒙药ZSP 的质量。
利益冲突所有作者均声明不存在利益冲突
