固-液-气三相环境下非均相苯加氢反应的原位核磁共振研究
2021-06-09高国梁王雪璐魏达秀姚叶锋
随 松,高国梁,王雪璐,魏达秀,姚叶锋
上海市磁共振重点实验室,华东师范大学 物理与材料科学学院,上海 200062
引 言
加氢反应是工业生产中的一类重要反应,例如用于提升木质素附加价值的苯酚加氢反应[1]、用于生产尼龙的苯加氢反应[2]、生产氢化可得松药品的反应,以及二氧化碳加氢还原反应等.因此,理解加氢反应机理和影响因素,对于提高反应物的利用率和催化剂效能具有重要的经济意义,同时也利于环境保护.目前,为了深入探究加氢反应机理,除了对反应前后的溶液进行常规检测外,研究者也开始使用一些原位或准原位技术[3-10]来观察加氢催化反应的中间产物,并期望通过这些方法来探测催化剂表面反应物分子之间的反应过程,最终验证猜想的理论,进而提出可能的加氢反应机理.据我们所知,固-液环境下的加氢反应往往存在固-液-气三相,这将大大增加原位检测的难度.目前大多数固-液加氢反应机理的研究都是在理想条件下(如气-固、超真空环境)完成的.然而,我们知道,反应环境对于反应路径及产物生成都起到非常重要的控制作用,因此开发能够在固-液-气三相共存体系进行原位检测的研究方法,并通过该方法在真实的三相反应条件下对加氢反应机理进行原位研究,将对加氢机理的认识起到非常重要的作用.然而,据我们所知,目前在真实固-液-气三相反应环境(溶液大量)且产物不分离的情况下对加氢反应机理进行原位在线的研究还尚未见报道.
在先前的研究[11]中,我们成功设计了可以在固-液环境下对光催化体系进行原位研究的核磁共振(NMR)设备,该设备可以在不分离产物的情况下对反应中间产物进行检测.随后,参照其他相关文献[12],经过设计改装,我们成功的将气体引入到反应器中,可以实现固-液-气三相环境下的原位检测.我们知道,对于非均相催化过程,催化转化率和产物选择性会受到催化剂纳米颗粒的大小和形状的影响[13-18].而且据报道,在1,3-丁二烯和氢气的加成反应中,研究发现负载只暴露(100)面的Pd四方体(Pd cube)纳米晶的共催化剂和负载只暴露(111)面的Pd八面体(Pd octa)纳米晶的共催化剂也会表现出不同的催化选择性.例如,1,3-丁二烯在Pd(100)上仅产生丁烯而不进一步加氢生成丁烷,并且暴露Pd(110)的共催化剂比暴露Pd(111)的共催化剂表现出对丁烯更高的选择性[19-21].与丁二烯的加氢反应类似,苯与氢气的加成反应也是催化剂结构敏感反应,但贵金属 Pd对于苯催化加氢反应影响的研究却很少.在苯的加氢反应中,苯在催化剂表面的吸附方式不同时,会按照不同的机理与氢气发生部分加成和完全加成反应[22].苯在催化剂表面的吸附方式与活性位点相关,分别通过π键[23,24]或π/σ键[25]与催化剂结合.而催化剂不同晶面上的活性位点不同,因此苯的加氢反应机理也可能不同.
本文使用原位NMR方法,在真实固-液-气三相共存体系(常温、2.5 bar压力条件,1 bar=105Pa)中,以商品钯纳米颗粒粉末(Pd NPs)研究压力、时间对非均相催化苯加氢反应的影响;以自制的暴露不同晶面的Pd cube/BCN、Pd octa/BCN共催化剂探究催化剂形貌对非均相催化苯加氢反应选择性的影响.
1 实验部分
1.1 实验试剂
聚氯乙烯吡咯烷酮(PVP,分子量为55 000)、四氯钯酸钾(K2PdCl4)、溴化钾(KBr)、L-抗坏血酸(C6H8O6)、柠檬酸、丙酮购自阿拉丁工业公司;氘代苯(C6D6)和Pd NPs购自Sigma-Aldrich公司;乙醇(C2H6O)和尿素购于国药集团化学试剂公司.药品未经进一步净化,直接使用.
块状g-C3N4(BCN)的制备:采用高温聚合法制备BCN.以尿素为前驱体,在马弗炉中以10 ℃/min的速率升温至550 ℃,并在此温度下保温3 h,随后自然降温至室温,得到淡黄色本体BCN.样品在使用前均进行干燥处理.
Pd/BCN的制备:第一步,采用简单的水相法制备Pd cube、Pd octa溶液.具体步骤如下:准确称量PVP(0.105 g)、KBr(0.300 g)和抗坏血酸(0.060 g),在室温下将其溶解于去离子水(8 mL)中,形成溶液A.将溶液A倒入三颈烧瓶(配有冷凝器和磁性搅拌子)中,在80 ℃油浴条件下加热10 min.同时称取0.065 g的K2PdCl4粉末,并将其溶解在去离子水(3 mL)中,形成溶液B.随后,将溶液B注入溶液A中并继续加热3 h.自然冷却后,将样品分散在纯水中,用高速离心机离心,并用丙酮和去离子水反复洗涤,制备出1 mg/mL的Pd cube溶液.Pd octa的制备与Pd cube相似,但以抗坏血酸和柠檬酸为还原剂,油浴温度为120 ℃.第二步,以BCN为载体,采用浸渍法将Pd负载在BCN上.具体方法如下:将80 mg的BCN粉末加入到坩埚中,随后向坩埚中加入适量Pd cube或Pd octa溶液,超声30 min.在90 ℃油浴条件下对坩埚进行加热,蒸发掉其中的水溶剂.最后,将坩埚置于马弗炉中,以10 ℃/min的速率升温至180 ℃并保温1 h,降温至室温后可以即得到不同形貌的Pd cube/BCN和Pd octa/BCN共催化剂,负载量分别为1%和0.61%.其中BCN作为Pd cube、Pd octa的载体,起到分散和固定的作用.样品在使用前均进行干燥处理.
1.2 仪器与参数
粉末X射线衍射(XRD)实验在Agilent 7890B型衍射仪上完成.测试条件:以20°/min的速度从5°扫描至90°.样品粉末事先研磨准备,所有XRD实验均在室温下进行.
使用扫描电子显微镜(SEM,Hitachi s-4800,Hitachi,Japan)和场发射透射电镜(TEM,Tecnai G2 F30,FEI,USA)对样品形貌进行检测.
使用图1所示的装置进行苯加氢反应的原位NMR监测.首先,将催化剂、氘代苯等先后加入到仿照Roth等[12]的方法特制的NMR样品管(S-5-600-SC-7,NORELL)中,并对其进行超声5 min处理,使催化剂在溶剂中充分分散;然后组装好装置,并将样品管放入NMR谱仪腔体中,通入氢气进行原位检测.压力表是VIGOUR品牌的VSR-1ES-200-10型,该表精度2.5级,通过压力表控制通入NMR样品管内氢气的压力.使用的NMR谱仪为Bruker 500 MHz型,探头为Bruker HXY三通道静态探头.质子共振频率为500.12 MHz,温度为298 K.1H NMR实验谱宽(SW)为12 ppm,脉冲宽度为4 μs(45°),采样时间为2 s,采样次数为1;13C NMR实验SW为238 ppm,采样时间为0.75 s,采样次数为5 000.所有实验以四甲基硅烷(TMS)进行定标(δH0.00,δC0.0).
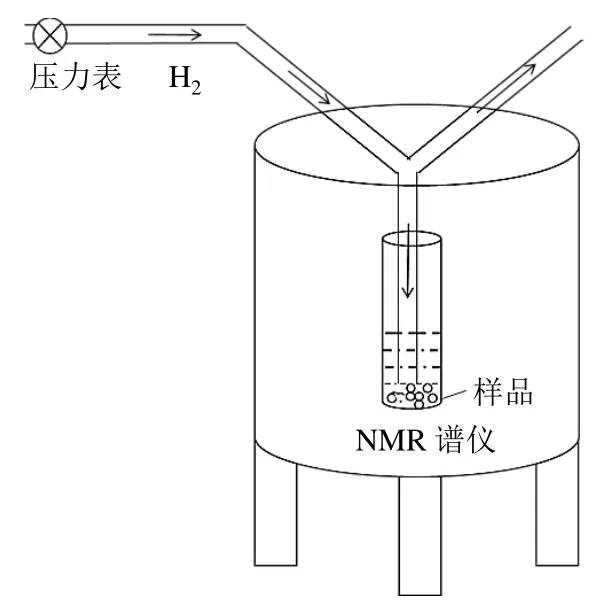
图1 原位NMR监测苯加氢反应的装置示意图Fig. 1 Schematic diagram for monitoring the reaction of benzene hydrogenation by operando NMR
2 结果与讨论
2.1 催化剂表征
图2为BCN、Pd NPs、Pd cube/BCN和Pd octa/BCN的XRD图谱.从图2可以看出,纯BCN样品中有两个衍射峰,位于27.5°和13.2°,分别对应于BCN的(002)和(100)晶面,符合BCN晶体结构.引入Pd cube、Pd octa后,Pd cube/BCN、Pd octa/BCN复合材料中仍然主要呈现BCN的特征峰,也可以看到位于 40.2°、46.2°、68.2°和 82.1°处 Pd(111)、(200)、(220)和(311)晶面的衍射峰,说明 Pd cube、Pd octa已经成功负载在BCN表面.

图2 Pd NPs、BCN和Pd/BCN复合材料的XRD图谱Fig. 2 XRD patterns of Pd NPs,BCN and Pd/BCN composites
随后,我们对所得的催化剂样品进行了SEM测试.如图3(a)所示,BCN是一种石墨状的透明层状结构;Pd cube则呈规则的立方体结构.

图3 (a) BCN和(b) Pd cube样品的SEM图像Fig. 3 SEM images of (a) BCN and (b) Pd cube
图4为Pd cube、Pd octa的TEM和高分辨率TEM(HRTEM)图像.从图4(a)和4(d)中可以看出,我们成功制备了不同晶面暴露的Pd cube、Pd octa.2个样品均具有很好的分散性,且未发现明显的局部堆积.如图4(b)所示,两个Pd晶格条纹之间的距离约为0.199 nm,符合面心立方Pd粉末衍射文件(PDF)卡片65-2867,该晶体的边缘晶格间距为0.195 nm,与面心立方(f.c.c)Pd晶体的(200)晶面间距一致[26],这一观察表明,我们成功地合成了(100)晶面暴露的Pd cube.由图4(e)所示的暴露(111)面的Pd octa的HRTEM图像可知,其晶格条纹间距为0.228 nm.通过对100~150个纳米颗粒的计算,我们得到了其粒径尺寸的大致分布.如图 4(c)和 4(f)所示,Pd cube的平均尺寸约为10.90 nm,Pd octa约为9.95 nm,两者相近.在尺寸相近的情况下,我们可以更好的对不同暴露面的Pd cube/BCN、Pd octa/BCN的催化性能进行考核.
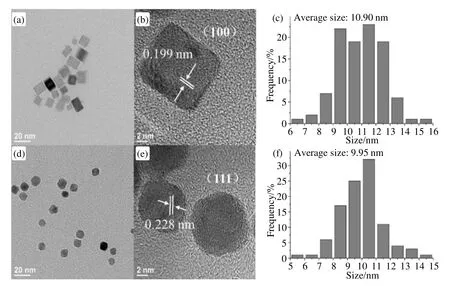
图4 Pd cube和Pd octa的TEM图像和HRTEM图像:(a) Pd cube的TEM图像;(b) Pd cube的HRTEM图像;(c) Pd cube的粒径分布图;(d) Pd octa的TEM图像;(e) Pd octa的HRTEM图像;(f) Pd octa的粒径分布图Fig. 4 TEM and HRTEM images of Pd cube and Pd octa. (a) TEM image of Pd cube; (b) HRTEM image of Pd cube;(c) Statistical graph of particle size distribution of Pd cube; (d) TEM image of Pd octa; (e) HRTEM image of Pd octa;(f) Statistical graph of particle size distribution of Pd octa
2.2 固-液-气三相加氢反应的原位NMR探究
观测和理解反应过程中出现的一些中间产物对于理解反应过程有重要意义[5,10].本文使用原位NMR技术监测了固-液-气三相共存体系中催化加氢反应的过程,并研究了反应压强、反应时间,以及催化剂表面性质对反应的影响.
2.2.1 1,3环己二烯的加氢反应过程的原位NMR监测
我们先使用Pd NPs作为催化剂,以苯加氢反应的中间产物之一——1,3-环己二烯的加氢反应作为探究体系来对加氢反应历程的监测进行初步尝试.我们将2 mg Pd NPs、15 μL 1,3-环己二烯和400 μL氘代苯溶剂先后加入NMR样品管中,然后对其进行超声处理5 min,使催化剂在溶剂中充分分散,随后向NMR样品管中通入氢气,并随着反应时间进行原位采样.图5(a)为1,3-环己二烯的加氢反应历程图(此反应中,相对于15 μL的1,3-环己二烯,溶剂与氢气反应可忽略不计),图5(b)为随着反应时间的增加,该体系的1H NMR谱图.从图5我们可以看出,未通入氢气之前,谱图中只有环己二烯的信号(A1、A2、A3).随着H2的加入,环己二烯转变为为环己烯(B1、B2、B3)和少量环己烷(C1).随着检测时间的增加,环己烯的产量出现先增加后减少的趋势,最后主要产物为环己烷.值得注意的是,根据相关文献[27],水在氘代苯中的化学位移为δH0.4左右;并且当苯环以π键方式吸附到催化剂表面,苯环上的质子可以与氘代水中的D原子互换,实际上这是生产氘代苯的方法之一[28].另外,在贵金属催化剂表面,吸附的重水分子与氢气解离产生的氢原子也会发生H和D互换[29],图5(b)水的信号峰增强也证明了这一点.因此,δH0.4为H2O/HDO的信号峰.
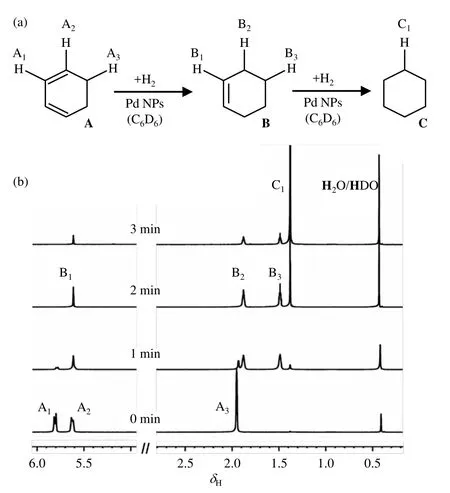
图5 (a) 1,3-环己二烯加氢过程;(b)不同反应时间采集的1H NMR谱.实验温度为298 K,压力为2.5 barFig. 5 (a) Hydrogenation of 1, 3-cyclohexadiene; (b)1H NMR spectra of 1,3-cyclohexadiene hydrogenation reaction,acquired at different reaction time. The experimental temperature is 298 K and the pressure is 2.5 bar
2.2.2 压力对非均相苯加氢催化反应的影响
我们选取商业购买的Pd NPs作为催化剂,研究了压力对非均相苯加氢催化反应的影响[如图6(a)所示].将2 mg Pd NPs和400 μL氘代苯先后加入NMR样品管中,然后对其进行超声处理5 min,使催化剂在溶剂中充分分散.随后向NMR样品管中通入不同压强的氢气,并在氢气通入3min后进行原位NMR采样,温度为298 K.如图6(c)所示,当向体系中通入氢气后,在δH1.38出现了明显单峰信号,我们将其归属为氘代环己烷(C6H6D6).为了进一步验证产物峰是环己烷,我们购买了环己烷标准物,并在相同的实验条件下测试了标准物的1H NMR谱图.如图6(b)所示,标准物与反应所得产物的1H NMR信号峰化学位移几乎相同.我们进一步做了一系列NMR实验对δH1.38处的峰进行归属,详细的归属过程见补充文件(扫描文章首页OSID码,或在文章网页版查看附件).从图6(c)中,我们可以看出,随着通气压力的增加,C6H6D6的产量呈现先下降后上升的趋势.在图 6(d)中,我们选择以C6H6的信号强度作为参考,之所以不选用水和TMS,而是C6H6作为该实验的内标,是因为在氘代苯中,苯分子绝大部分以C6D6的形式存在,只有很少以C6H6形式存在,所以与氢气发生加成反应的大都是C6D6分子,而C6H6的信号强度基本不变;同时,水和TMS会随着氢气的通入而发生信号强度的改变.并且,我们首先通过配制一系列浓度的非氘代苯C6H6的标准溶液,对反应体系中C6H6的浓度进行了标定.在配制标准溶液过程中,我们取用一定体积非氘代苯,然后用氘代苯将其稀释十倍,它们的体积比接近1:9,然后取出一定体积的此溶液,进一步稀释,多次稀释操作后得到一系列浓度梯度的溶液.图6(d)中的以C6H6D6相对于C6H6的积分强度推算出的浓度为纵坐标,以通入氢气的压强为横坐标作图.计算过程的误差主要来源于各信号积分误差(约为5%).从图6(d)可以看出,在我们实验的五个压力值范围内,当通气压强在4 bar时,苯加氢反应的产率最高.
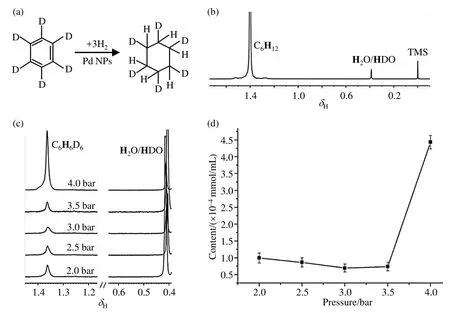
图6 (a)氘代苯加氢反应示意图;(b)环己烷标准品的1H NMR谱图;(c)通气压力对苯加氢催化反应影响的1H NMR谱图;(d)不同压力下苯加氢反应产物C6H6D6的含量.实验温度为298 KFig. 6 (a) The schematic diagram of deuterated benzene hydrogenation reaction; (b)1H NMR spectrum of cyclohexane standard;(c) Influence of pressure on heterogeneous benzene hydrogenation reaction; (d) The trends of C6H6D6 of benzene hydrogenation products at different pressures. The experimental temperature is 298 K
2.2.3 通气时间对非均相苯加氢催化反应的影响
随后,我们研究了通气时间对非均相苯加氢催化反应的影响.反应体系为2 mg Pd NPs和400 μL氘代苯,温度为298 K.通气压力为2.5 bar,通入时间分别为2、4、6和8 min.图7为不同通气时间的苯加氢产物C6H6D6的1H NMR谱图.可以看出,产物C6H6D6的产量与通入氢气的时间呈正相关关系.从图7(a)和7(b)可以看出,随着通入氢气时间的延长,C6H6D6的产率逐渐增强.这可能是由于氢气在溶液中需要经历迁移吸附和裂解过程,因此,通入时间越长,到达催化剂表面的氢气越多.也就是说,在反应初始阶段,氢气在催化剂表面的吸附、裂解过程为加氢反应的决速步骤.图7(b)依旧是以苯(C6H6)的信号强度作为参考.
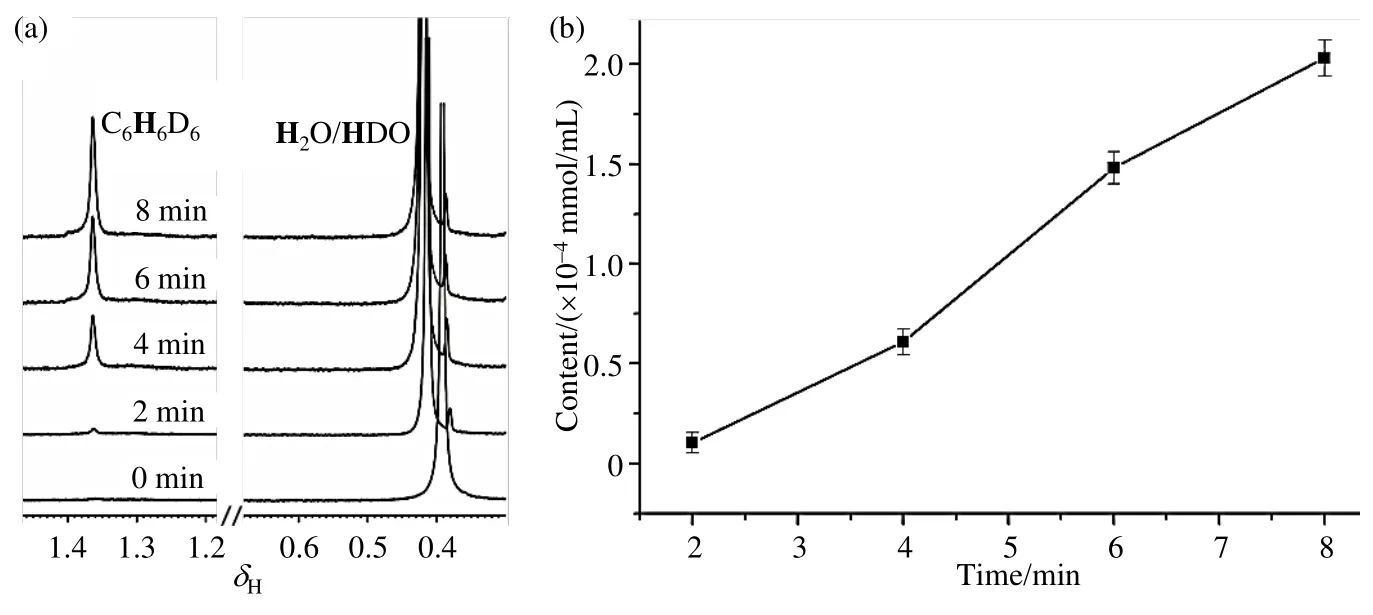
图7 (a)通气时间对苯加氢催化反应影响的1H NMR谱图;(b)不同反应时间的苯加氢产物C6H6D6的含量.实验温度为298 K,实验压力为2.5 barFig. 7 (a) 1H NMR spectra of heterogeneous benzene hydrogenation reaction, acquired at different reaction time; (b) Influence of reaction time on the trends of C6H6D6 of benzene hydrogenation reaction. The experimental temperature is 298 K and the pressure is 2.5 bar
2.2.4 催化剂表面性质对非均相苯加氢催化反应的影响
在非均相催化加氢反应中,催化剂的表面性质对于催化反应往往起到非常重要的作用.接下来,我们对具有不同暴露晶面的Pd cube/BCN、Pd octa/BCN催化剂对苯加氢反应的选择性进行了研究.我们观察到,当氢气通入NMR样品管后,Pd NPs会出现团聚现象,比表面积减小,催化效果减弱.因此,为了提高Pd的利用率,同时探究哪个晶面对产物具备更好的选择性,我们尝试用负载的方法将Pd cube、Pd octa均匀负载在没有苯加氢性能的载体上,以其起到分散的作用.我们选取了能在溶液中较好分散的BCN作为负载的载体,并将Pd cube、Pd octa负载到BCN上,同时利用Pd cube、Pd octa分别只暴露(100)面和(111)面的结构来研究不同暴露晶面的催化剂对苯加氢反应的选择性.反应体系为2 mg催化剂(BCN、Pd cube/BCN及Pd octa/BCN)和400 μL氘代苯,温度为298 K.如图8所示,在2.5 bar压力条件下通入氢气4 min后检测发现,以Pd cube/BCN为催化剂的体系没有任何苯加氢的产物,这可能是由于负载Pd含量较低的原因.而在Pd octa/BCN样品的1H NMR谱中则出现了位于δH1.48的环己烯中远离双键的质子的信号.为了验证产物峰是环己烯,我们购买了环己烯标准物,并在相同的实验条件下测试了标准物的1H NMR谱图.如图8(b)所示,标准物与反应所得产物的1H NMR信号峰化学位移几乎相同.通过对比,可以得出暴露(111)面的Pd octa/BCN催化剂对于苯环加氢具有催化效果.也就是说不同暴露晶面的催化剂对苯加氢反应具有选择性,如图8(c)所示.
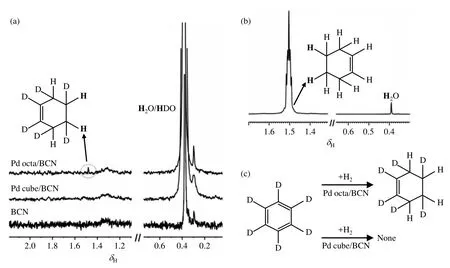
图8 (a) BCN、Pd cube/BCN和Pd octa/BCN催化苯加氢反应体系的1H NMR谱图.实验温度为298 K.实验压力为2.5 bar;(b) 环己烯标准物中远离双键的质子的1H NMR谱图;(c)不同暴露晶面的催化剂选择性催化苯加氢反应示意图Fig. 8 1H NMR spectra of catalytic hydrogenation of benzene, catalyzed by BCN, Pd cube/BCN and Pd octa/BCN. The experimental temperature is 298 K and the reaction pressure is 2.5 bar; (b) 1H NMR spectrum of cyclohexene standard material away from carbon-carbon double-bond; (c) Schematic diagram of selective hydrogenation of benzene catalyst by nanoparticles with different exposed surfaces
苯在催化剂上的吸附方式不同,会按照不同的机理与氢气发生部分加成和完全加成反应.苯在催化剂表面的吸附方式与活性位点相关,分别以π键、以及π/σ键与催化剂结合.因此,由于在不同晶面上活性位点的分布不同,苯的加氢反应产物也会有所差别[30-33].本文使用不同晶面暴露的 Pd cube/BCN、Pd octa/BCN为共催化剂,在负载量约为1 %的条件下,进一步证实了这一点,对比于Pd(100)晶面,Pd(111)晶面将利于产生环己烯.在 Pd(111)晶面上,苯环上的四个碳原子与活性位点中的金属原子以π键吸附,另外两个碳原子以σ键吸附;但只有以π键吸附的原子才有反应活性,σ键吸附则没有.在Pd(100)晶面上,苯环六个碳原子的两种吸附方式则与Pd(111)面相反,所以其催化活性较低.然而,由于苯环分子与四个氢原子发生加成反应,π键相互作用减弱,导致产物环己烯从催化剂表面脱附无法进一步加氢,因此没有产物环己烷.此外,不同晶面对反应产物选择性和活性的影响或许还与晶面和物质的结合能有关.据我们所知,通常情况下苯分子在(111)面的活化能要比(100)面小,所以(111)暴露面为主的Pd octa催化剂在苯加氢反应中具有更强的催化活性.这可能是由于不同形貌纳米颗粒的边角原子所占比例不同导致[34,35].在不同形貌的催化剂上,边原子属于弱配位原子,对反应物的吸附能力较弱;并且相比较于面原子,它的催化活性也较低.纳米颗粒尺寸相近的条件下,边原子在Pd octa中的占比高于其在Pd cube中的占比;所以产物环己烯更易脱附,环己烯也更易形成.同时,在反应物浓度较大的情况下,反应物分子在边原子上的吸附会锁住面原子对反应物的吸附,导致催化活性位点失活.在我们的反应体系中,液态的苯溶剂直接包裹住催化剂,可能是由于边原子的吸附导致催化活性较低,所以环己烯的信号峰不强.
3 结论
我们使用原位NMR方法对固-液-气非均相催化环境下的苯加氢反应机理进行了研究.以Pd NPs、和不同形貌的Pd cube/BCN、Pd octa/BCN作为研究对象,系统研究了反应压强、反应时间,以及催化剂表面性质对加氢反应的影响.在反应进行中,无需分离产物,对体系真实状态下的过程实现检测.结果表明,压强和反应时间都可以影响加氢反应产物,不同的催化剂对苯与氢气的加成反应催化效果不同.在研究的压强条件下(2.0 bar、2.5 bar、3.0 bar、3.5 bar和4.0 bar),苯加氢反应的产率在4.0 bar时表现出最高.在通气时间方面,在反应起始阶段,随着通入氢气时间的延长,苯加氢反应的产率逐渐增强.催化剂表面性质方面,不同晶面暴露的Pd共催化剂苯加氢产物不同.通气时间为4 min,2.5 bar压力条件下,对比于以(100)晶面为主的Pd cube/BCN,以(111)暴露面为主的Pd octa/BCN对苯的加氢反应更倾向于产生环己烯.该原位 NMR方法为研究固-液-气催化环境下的加氢反应机理提供了机会.
利益冲突
无
附件材料
“产物环己烷信号的归属”
