4例自身免疫性胶质纤维酸性蛋白星形胶质细胞病的临床特征分析
2021-06-07郭昆典洪桢
郭昆典 洪桢
自身免疫性胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)星形胶质细胞病是2016年由Fang等[1]提出的一种针对中枢神经系统的炎性自身免疫性疾病,目前认为GFAP免疫球蛋白G(GFAP-IgG)是诊断该病的特异性生物标志物。该病临床罕见,目前全球仅有300余例报道,我国南方地区有少量病例报道[2-3],西部地区尚缺乏报道。该研究首次对中国西部地区4例自身免疫性GFAP星形胶质细胞病的临床特征进行回顾性总结,旨在提高临床对该病的认识,为其诊治提供参考。
1 对象和方法
1.1 对象回顾性收集2019年6月1日至2020年11月31日由四川大学华西医院收治的确诊自身免疫性GFAP星形胶质细胞病患者4例。患者符合如下纳入标准[1]:(1)临床表现符合脑炎、脑膜炎、脊髓炎或上述综合征的组合;(2)脑脊液GFAP-IgG阳性。
1.2 方法
1.2.1资料收集:收集并分析患者的人口学特点、急性期临床表现、实验室检查、影像学检查、神经电生理检查、治疗等临床资料。
1.2.2抗体检测:采用IgG抗体检测试剂盒(德国,Euroimmun)通过基于细胞底物(cell based assay,CBA)的间接免疫荧光法对患者血清及脑脊液的自身免疫性脑炎抗体谱、抗GFAP抗体、水通道蛋白4(aquaporin 4,AQP4)抗体和髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体进行检测;采用IgG抗体检测试剂盒(德国,Euroimmun)通过免疫印迹法对患者血清及脑脊液神经副肿瘤抗体谱进行检测。所有检测均由作者团队自行完成。
1.2.3预后评估:通过标准化问卷及定期门诊随访获取患者预后情况,采用改良Rankin量表(modified Rankin scales,mRS)在症状高峰期、免疫治疗1个月及末次随访时对患者症状的严重程度及功能预后进行评估:0分指完全无症状;1分指尽管有症状但未见明显残疾,能完成所有日常工作和生活;2分指轻度残疾,不能完成病前所能从事的活动,但不需帮助即可处理自己的日常生活;3分指中度残疾,需部分帮助,但能独立行走;4分指中重度残疾,不能独立行走,需他人帮助处理日常事务;5分指重度残疾,卧床,二便失禁,日常生活完全依赖他人;6分指死亡。
2 结果
2.1 临床特征结果见表1。患者中男和女各2例,均为急性或亚急性起病,临床表现均符合脑膜脑炎或脑膜脑脊髓炎。有3例存在前驱感染可能,均表现为发热。首发症状以头痛最为常见,此外也可出现肢体无力和精神行为异常。所有患者急性期均出现脑膜症状,表现为颈强直,其中3例伴头痛;4例均出现精神障碍;3例出现不同程度的认知功能下降和意识障碍;2例出现痫性发作,均为部分性发作;2例出现肢体无力,其中1例合并周围神经病;2例出现自主神经功能障碍,表现为肠麻痹。此外,脑干损害、震颤、视物模糊及抗利尿激素分泌异常综合征各见于1例患者。
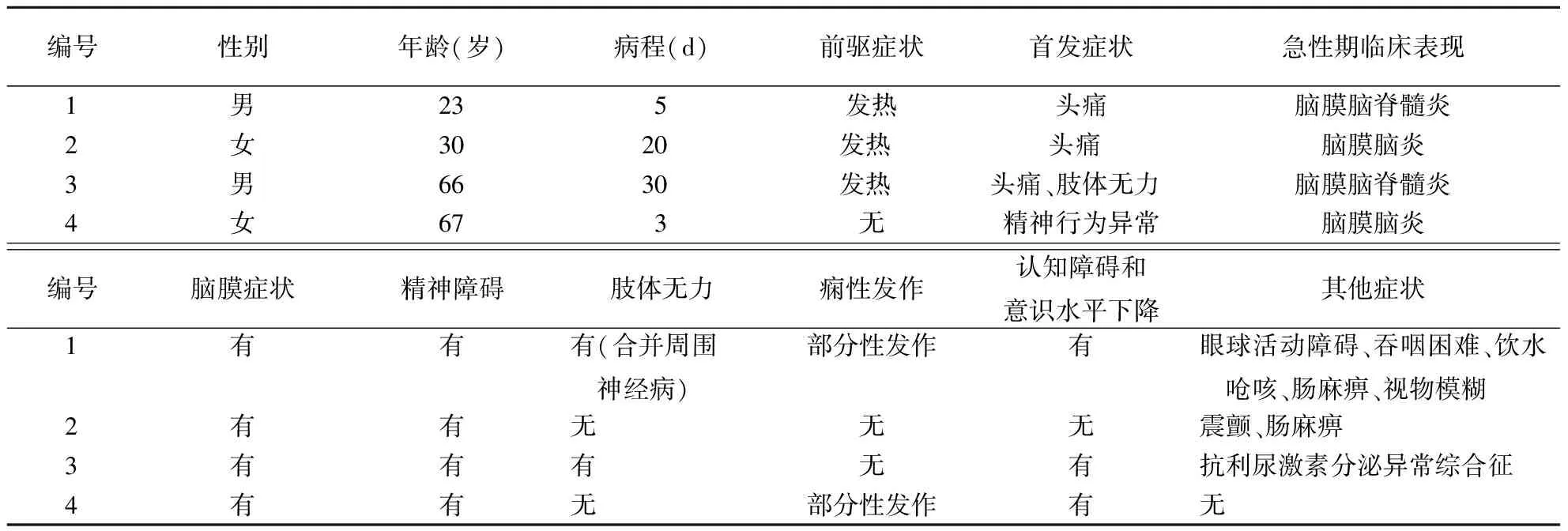
表1 4例自身免疫性GFAP星形胶质细胞病患者的临床特征
2.2 实验室检查(1)脑脊液检查:脑脊液压力升高2例,有核细胞数升高4例〔正常参考值(0~5)×106/L〕,均以淋巴细胞为主;蛋白升高3例(正常参考值0.15~0.45 g/L),氯化物降低3例(正常参考值120~130 mmol/L),葡萄糖降低2例(正常参考值2.5~4.4 mmol/L)。脑脊液病原学检查(涂片、培养、结核杆菌抗体及DNA、病毒PCR)均未见异常。(2)同步血生化检查:低钠血症2例(正常参考值135~145 mmol/L)。(3)抗体检测:4例患者脑脊液抗GFAP抗体均阳性,2例血清抗GFAP抗体阳性。患者1合并血清抗Yo抗体阳性,患者4合并血清抗Hu抗体阳性。2例合并脊髓炎的患者行血清及脑脊液MOG和AQP4抗体检测,结果均阴性。所有患者γ干扰素释放结合感染T细胞斑点试验、PPD皮试、血清肿瘤标志物和自身抗体全套(抗核抗体、抗双链DNA抗体、抗核内可溶性抗原抗体、抗Sm抗体、抗核糖核蛋白体抗体、抗SSA抗体、抗SSB抗体及抗Scl-70抗体)均未见异常。结果见表2。
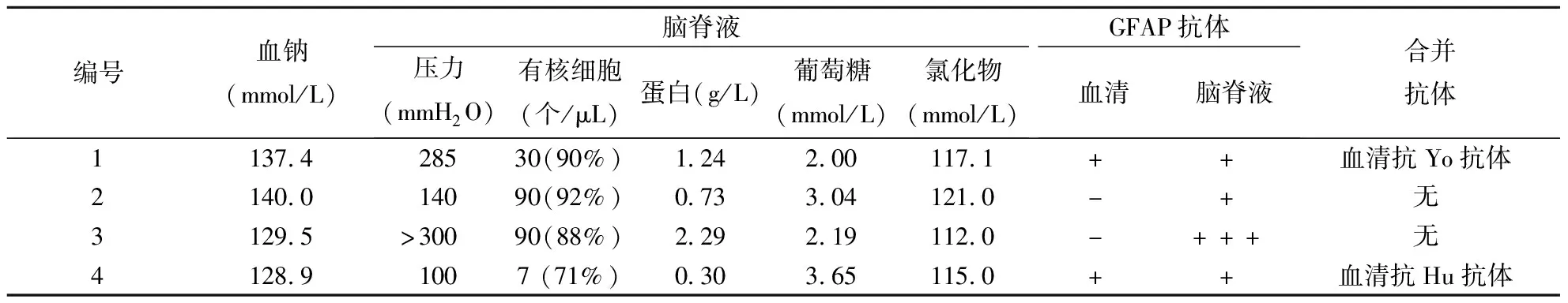
表2 4例自身免疫性GFAP星形胶质细胞病患者实验室检查结果
2.3 影像学表现结果见表3。4例患者头颅MRI均出现颅内多发异常信号,病灶累及皮层及皮层下、侧脑室周围白质、基底节区、胼胝体、小脑半球等,表现为长T1或等T1、长T2信号,Flair像呈高信号,部分病灶增强像可见点线状强化,其中患者3矢状位可见侧脑室周围放射状线样强化(图1)。2例行颈、胸、腰段脊髓MRI,其中患者1见颈段脊髓长T1长T2信号(图2A),Flair像呈高信号,增强像可见软脊膜和颈段脊髓部分病灶线样强化(图2B)。患者2脊髓MRI未见明显异常。所有患者经胸部、全腹增强CT及泌尿系彩超筛查肿瘤(女性增加妇科筛查),均未见占位性病变。

注:B、D分别为图A、C方框部位放大图
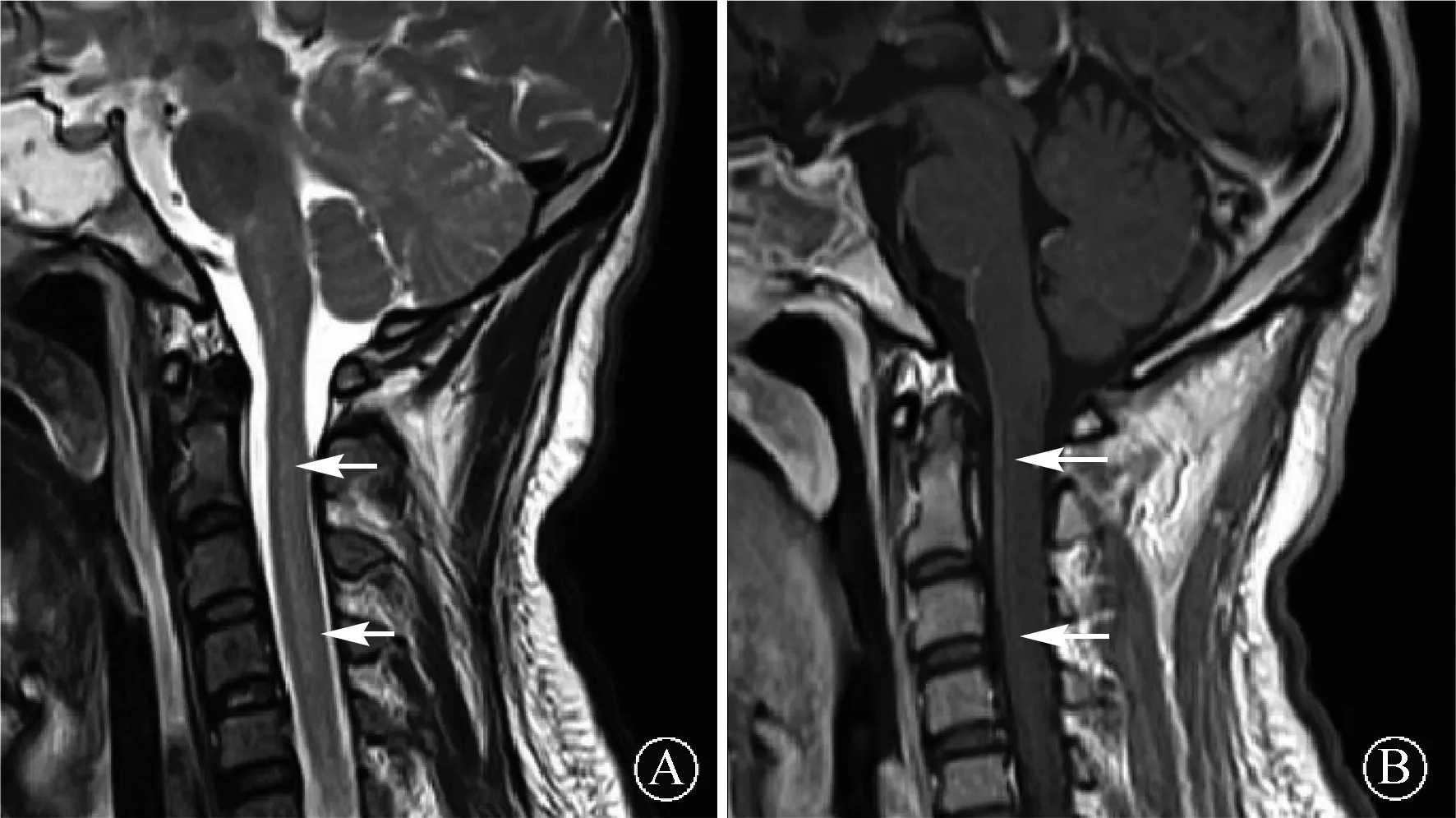
图2 患者1颈椎MRI检查显示颈段脊髓长节段长T2信号(A,箭头所示),增强像见软脊膜和脊髓部分病灶线样强化(B,箭头所示)
2.4 电生理检查常规脑电图检查显示1例出现局灶性慢波,3例出现弥漫性慢波(表3)。患者1因查体发现双侧肱二头肌反射、肱三头肌反射、桡骨膜反射和膝反射活跃,双侧踝反射消失,双下肢肌力明显弱于双上肢,考虑合并周围神经损害,经肌电图检查发现右侧腓总神经运动传导复合肌肉动作电位(CMAP)波幅降低,左侧腓总神经运动传导未引出明显波形,双侧腓总神经、左侧胫神经F波未引出,证实双下肢呈神经源性损害表现。此外,患者1由于视物模糊行视觉诱发电位检查,结果提示双侧P100波潜伏期延长,波幅降低,该患者眼底照相未见明显异常。

表3 4例自身免疫性GFAP星形胶质细胞病患者辅助检查结果
2.5 治疗及转归所有患者于急性期接受糖皮质激素冲击或联合静脉丙种球蛋白治疗,均对免疫治疗有反应,mRS评分降低。4例患者出院后均口服激素维持治疗,维持治疗中位时间为6个月。患者1和患者4口服抗癫痫药(anti-epileptic drugs,AEDs)治疗,未再有痫性发作。随访3~17个月,中位随访时间13个月,4例患者末次随访mRS评分均小于3分,无复发。患者1和患者2分别于入院后4个月和2个月复查血清及脑脊液抗GFAP抗体均阴性,MRI提示病灶缩小且未见异常强化。结果见表4。

表4 4例自身免疫性GFAP星形胶质细胞病患者治疗及临床转归
3 讨论
GFAP是成熟星形胶质细胞主要的中间丝蛋白,参与包括神经再生、突触可塑性以及反应性胶质增生在内的多种功能[4]。有学者推测GFAP星形细胞病是由细胞毒性T细胞介导的免疫反应所致,而抗GFAP抗体为这一免疫反应的标志物[5]。尽管病毒感染及肿瘤可能作为触发该免疫反应的因素,但具体发病机制仍需进一步研究[6]。
自身免疫性GFAP星形胶质细胞病可于各年龄段发病,男女比例接近。约40%的患者发病前出现非特异性感染症状。该病主要表现为急性或亚急性的脑膜炎、脑炎、脊髓炎或上述炎症的组合(下文统称脑膜脑脊髓炎表型),以脑膜脑炎最为常见,孤立的脊髓炎罕见[5]。头痛、颈强直、恶心等脑膜症状见于半数患者,其他常见的临床表现包括意识障碍、震颤、精神症状和痫性发作等,约30%的患者可出现视物模糊,少数患者还可出现共济失调、自主神经功能障碍、周围神经病等症状[5]。该研究中4例患者均符合脑膜脑脊髓炎表型,临床症状与既往的报道相符。
大多数患者行头部MRI检查可发现异常,病灶累及大脑皮质、基底节区、下丘脑、脑室周围白质、小脑、脑干、脑膜以及颅骨等[2,5]。约半数患者在头部增强MRI检查中可发现侧脑室周围线样放射状血管周围强化,这被认为是本病最具特异性的影像学表现,类似的放射状强化也可出现在小脑。脊髓MRI主要表现为长节段的异常信号,常累及中央灰质,部分病例可见强化[6-7]。该研究中4例患者影像学特点均符合文献报道,头部MRI检查均提示颅内多发异常信号。
患者脑脊液多呈炎性改变,约90%患者出现以淋巴细胞为主的有核细胞数增高,80%出现蛋白质增高,半数以上存在寡克隆带,部分患者IgG指数可增高[5]。此外,脑脊液中葡萄糖可出现轻中度下降。鉴于脑脊液特点及临床表现,不少患者可能在该疾病早期被误诊为结核性脑膜(脑)炎[8-11]。事实上,患者3在早期曾被怀疑结核性脑膜脑炎,并接受诊断性抗结核治疗。
目前普遍认为,脑脊液GFAP-IgG对脑膜脑脊髓炎表型具有较高的特异性和敏感性,是诊断自身免疫性GFAP星形胶质细胞病的标志物,但仅血清抗GFAP抗体阳性的患者临床表现具有异质性[7]。近来Paul等[12]提出血清抗GFAP抗体可能参与免疫介导的周围神经病。因此,该病的临床范围是否更广及血清抗GFAP抗体对该病的诊断价值仍有待商榷,尚缺乏统一的诊断标准[9]。
抗GFAP抗体阳性的患者合并其他神经系统自身抗体的情况并不少见,其中以抗NMDAR抗体最为常见,AQP4抗体次之[5]。Yang等[13]研究表明除发病年龄外,患者是否合并其他自身抗体在临床特征上并无明显差异。该研究中患者1和患者4分别于血清中发现抗Yo抗体和抗Hu抗体,但2例患者的临床表现均与自身免疫性GFAP星形胶质细胞病相符,且均未发现肿瘤。鉴于该2例患者仅在血清中发现低滴度的神经副肿瘤抗体且未经CBA法及基于组织底物的间接免疫荧光法进一步验证,尚不能排除假阳性的可能。
自身免疫性GFAP星形胶质细胞病患者脑电图检查可发现非特异性的改变,主要表现为弥漫性慢波[8],仅有1例儿童出现抗NMDAR抗体脑炎特征性“δ刷”的报道[14]。该研究中患者1脑电图检查发现局灶性慢波,并在病程中出现痫性发作,其余3例均为弥漫性慢波。此外,患者视觉诱发电位检查可见异常,眼科检查可发现双侧对称的视盘水肿,光学相干断层扫描可显示视网膜神经纤维层增厚。多数患者无视觉症状,少数患者视野检查可发现轻度弓形视野缺损或盲点扩大,但严重视力损害罕见[2,15]。该研究中患者1急性期视觉诱发电位检查提示双侧P100波潜伏期延长,波幅降低,但眼底检查未发现视盘水肿。
自身免疫性GFAP星形胶质细胞病尚无指南明确治疗方案,目前应用最广的急性期治疗为大剂量类固醇激素冲击,其次是静脉注射丙种球蛋白和血浆置换,维持治疗可选择口服泼尼松并缓慢减量[1,2,7,9]。约20%~50%的患者可复发,主要发生在口服激素减量过程中[1,5,7],此时可考虑加用免疫抑制剂如吗替麦考酚酯或硫唑嘌呤,也可选用利妥昔单抗或环磷酰胺[5]。患者的长期预后差别较大,有国内学者认为多数患者遗留残疾且长期预后不佳[2-3]。该研究中4例患者均对急性期免疫治疗反应良好,但患者1遗留视物模糊及夜间遗尿,患者3遗留肢体无力。
约25%自身免疫性GFAP星形胶质细胞病患者可在发病之后发现肿瘤,其中卵巢畸胎瘤约占75%[5]。因此,对该病患者应进行积极的肿瘤筛查并长期随访。
综上所述,自身免疫性GFAP星形胶质细胞病以急性或亚急性起病的脑膜炎、脑炎、脊髓炎或上述综合征的组合为主要表现。当临床上出现急性或亚急性起病且以脑膜脑脊髓炎相关症状为主要临床表现,尤其脑脊液检查发现淋巴细胞升高或头部MRI检查发现脑室周围放射状线样强化的患者,应尽快完善脑脊液和血清抗GFAP抗体的检测以明确诊断,并及时予以免疫治疗,以改善症状及预后。
