一测多评法测定不同产地巴戟天中4种环烯醚萜含量
2021-06-07王丽丽崔庆艳张素中王永生王诗涵
王丽丽,崔庆艳,张素中,王永生,王诗涵
1中山大学新华学院 药学院,广州 510520;2吉林大学 药学院,长春 130021;3吉林农业大学 中药材学院,长春 130118
巴戟天来源于茜草科(Rubiaceae)植物巴戟天(MorindaofficinalisHow.)的干燥根[1],该中药材始载于《神农本草经》,列为上品,历代本草都有记载,是著名的保健中药材,也是我国“四大南药”之一,以广东为道地产区,具有抗骨质疏松、补肾阳、抗氧化、抗肿瘤、抗炎、抗疲劳、增强机体免疫力等作用[2,3]。巴戟天中主要成分有环烯醚萜类、蒽醌类、糖类及有机酸类等[3,4]。以水晶兰苷为代表的环烯醚萜类成分具有抗炎、镇痛、抗氧化、抗肿瘤、抗衰老、抗肥胖等作用[4-8]。车叶草苷还具有骨保护作用和降低急性肺损伤的作用[9]。
近年来,随着人们的意识以医疗为重点转向以预防保健为重点,巴戟天的需求量不断提升,逐渐出现了以次充好的现象。《中国药典》2015年版巴戟天含量测定项选用耐斯糖一种成分作为定量指标[1],存在一定的局限性,若能将多个指标性成分同时纳入定量指标,将对巴戟天的品质会有更全面的评估。
一测多评法是利用中药材有效成分之间的内在函数和比例关系,通过测定一种成分(对照品易于获得)而实现多种成分(对照品难以获得或价格昂贵)的同步测定,该方法是《中国药典》2015版重点推广的方法,现已成为中药材多指标成分含量测定的首选方法[10]。但当前国内外尚无采用一测多评法测定其含量的报道。
本实验建立一测多评法,首次实现对巴戟天中4种环烯醚萜类成分水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸、车叶草苷的同步测定,并对不同产地巴戟天进行质量控制,为其质量标准修订提供参考。
1 仪器和试药
Agilent 1260 型液相色谱仪(安捷伦公司);Waters Alliance E2695型液相色谱仪(沃特世公司);LC-10ATVP型岛津液相色谱仪(产地:日本);色谱柱:Venusil MP C18色谱柱(250 mm×4.6 mm,5 μm)、Agilent ZORBAX SB-Aq色谱柱(250 mm×4.6 mm,5 μm);Sartorious型十万分之一天平(北京赛多利斯科学仪器有限公司);WP-UP-Ⅲ-10型超纯水机(四川沃特尔水处理设备有限公司)。
水晶兰苷(批号:5945-50-6,成都埃法生物科技有限公司);去乙酰基车叶草苷酸(批号:14259-55-3,成都埃法生物科技有限公司);车叶草苷酸(批号:25368-11-0,成都埃法生物科技有限公司);车叶草苷(批号:14259-45-1,成都埃法生物科技有限公司);11批巴戟天样品来源信息见表1,经吉林大学王广树教授鉴定,为茜草科(Rubiaceae)植物巴戟天(MorindaofficinalisHow.)的干燥根;乙腈为色谱纯(美国Fisher公司);水为超纯水(自制);其余试剂均为分析纯。

表1 11批巴戟天样品来源信息Table 1 Eleven batches of M.officinalis source information
2 方法与结果
2.1 色谱条件
采用Venusil MP C18色谱柱(250 mm×4.6 mm,5 μm);流动相为:乙腈(A)-0.2%磷酸溶液+0.01 mol/L磷酸氢二钾缓冲盐(B),梯度洗脱(0~12 min,1% A;12~30 min,1%A→17% A;30~40 min,17% A);流速为1.0 mL/min;检测波长为235 nm;柱温为25 ℃;进样量为20 μL。
2.2 溶液的制备
2.2.1 混合对照品溶液的制备
精密称取水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸和车叶草苷对照品适量,制成质量浓度分别为2.540 0、0.840 0、5.025 0×10-2、2.028 0×10-2mg/mL的混合对照品储备液。精密量取混合对照品储备液适量,制成质量浓度依次为0.635 0、0.210 0、1.256 2×10-2、5.070 0×10-3mg/mL的混合对照品溶液。
2.2.2 供试品溶液的制备
称取巴戟天药材粉末0.5 g,精密称定,置具塞锥形瓶中,加入80% 的甲醇,超声提取30 min(功率为400 W,频率为90 Hz),过滤,滤渣再同法提取一次,合并提取液,水浴蒸干,用初始流动相溶解。过滤,取续滤液,即得。
2.3 系统适用性试验
分别取“2.2”项下混合对照品溶液和供试品溶液适量,按“2.1”项下色谱条件进样测定,记录色谱图。结果,混合对照品溶液与供试品溶液在相同时间处均有相应的色谱峰出现,各色谱峰分离较好,分离度均大于1.5;空白溶剂在相应保留时间处没有色谱峰出现,对测定无干扰;理论塔板数以所测目标成分计均大于8 000(见图1)。
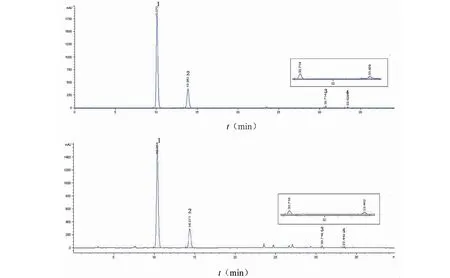
图1 混合对照品溶液(A)和样品(B)HPLC图谱Fig.1 HPLC of reference substances(A)and samples(B)注:1:水晶兰苷;2:去乙酰基车叶草苷酸;3:车叶草苷酸;4:车叶草苷。Note:1:Monotropein;2:Deacetyl asperulosidic acid;3:Asperulosidic acid;4:Asperuloside.
2.4 线性关系考察
精密量取“2.2.1”项下混合对照品储备液1.25、2.50、3.75、5.00、7.50 mL,置于20 mL量瓶中,加入初始流动相定容。按“2.1”项下色谱条件进行测定,记录峰面积,以4种成分的质量浓度(X,μg/mL)为横坐标,峰面积(Y)为纵坐标,进行线性回归,结果见表2。

表2 4种环烯醚萜成分的线性关系考察Table 2 Investigation on the linear relationship of four kinds of iridoids
2.5 精密度试验
取“2.2.1”项下混合对照品溶液,按“2.1”项下色谱条件连续进样测定6次,记录峰面积。结果,水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸、车叶草苷峰面积的RSD分别为0.17%、0.08%、0.36%、0.47%(n=6),表明仪器精密度良好。
2.6 稳定性试验
取“2.2.2”项下供试品溶液适量,分别于室温下放置0、2、4、6、8、10、12、24 h,按“2.1”项下色谱条件进样测定,记录峰面积。结果,水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸、车叶草苷峰面积的RSD值分别为0.97%、0.43%、0.94%、1.31%(n=6),表明供试品溶液于室温下放置24 h内稳定性良好。
2.7 重复性试验
取同一批巴戟天药材粉末,按“2.2.2”项下方法平行制备6份供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积,并按ESM法计算4种成分的含量。结果,水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸、车叶草苷的平均含量分别为12.02、3.85、0.12、0.08 mg/g,RSD分别为0.74%、0.80%、1.08%、1.34%(n=6),表明本方法重复性良好。
2.8 加样回收率试验
取同一批巴戟天药材粉末6份,每份约0.25 g,精密称定,按样品含量-对照品约(1∶1)的比例加入一定量对照品,按“2.2.2”项下方法平行制备6份供试品溶液,再按“2.1”项下色谱条件进样测定,记录峰面积并计算加样回收率。结果,水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸、车叶草苷的回收率分别为100.03%、99.97%、99.35%、99.68%,RSD分别为0.32%、1.05%、0.93%、1.01%(n=6),表明该方法的准确度较好。
2.9 相对校正因子的测定
取“2.2.1”项下混合对照品溶液5、10、15、20、30 μL,按“2.1”项下色谱条件进样测定,记录峰面积。以去乙酰基车叶草苷酸(Ⅱ)为内参物,根据公式按公式fk/m=fk/fm=Wk×Am/(Wm×Ak)[11],式中Ak为内参物峰面积,Wk为内参物的质量或浓度,Am为其他组分m的峰面积,Wm为其他组分的质量或浓度。以去乙酰基车叶草苷酸(Ⅱ)为内参物,分别计算水晶兰苷(Ⅰ)、车叶草苷酸(Ⅲ)、车叶草苷(Ⅳ)的相对校正因子(见表3)。

表3 巴戟天中3种环烯醚萜类成分相对校正因子Table 3 Relative correction factors of three iridoids in M.officinalis
2.9.1 不同仪器及色谱柱对相对校正因子的影响
考察三种不同型号仪器Agilent 1260、Waters E2695、LC-10ATVP液相色谱仪和两种不同色谱柱Venusil MP C18和Agilent ZORBAX SB-Aq(均为250 mm×4.6 mm,5 μm)对相对校正因子的影响,分别计算各成分的相对校正因子及RSD值。结果表明,使用不同仪器和色谱柱,各成分相对校正因子的RSD值均小于2.00%(见表4)。

表4 不同仪器和色谱柱对相对校正因子的影响Table 4 Effect of different instruments and columns on relative correction factors
2.9.2 不同实验人员对相对校正因子的影响
考察了3名实验人员对各成分相对校正因子的影响,各成分相对校正因子的RSD值均小于2.00%(见表5)。

表5 不同实验人员对相对校正因子的影响Table 5 Effect of different experimenter on relative correction factors
2.9.3 待测成分色谱峰的定位
采用相对保留时间定位法,考察其在3个高效液相色谱色谱仪、2个不同色谱柱上各待测组分色谱峰的相对保留时间(t)和重现性。结果表明,在相同的色谱条件下,各成分相对保留时间在不同仪器和色谱柱条件下变化较小,RSD值均小于4.00%(见表6)。
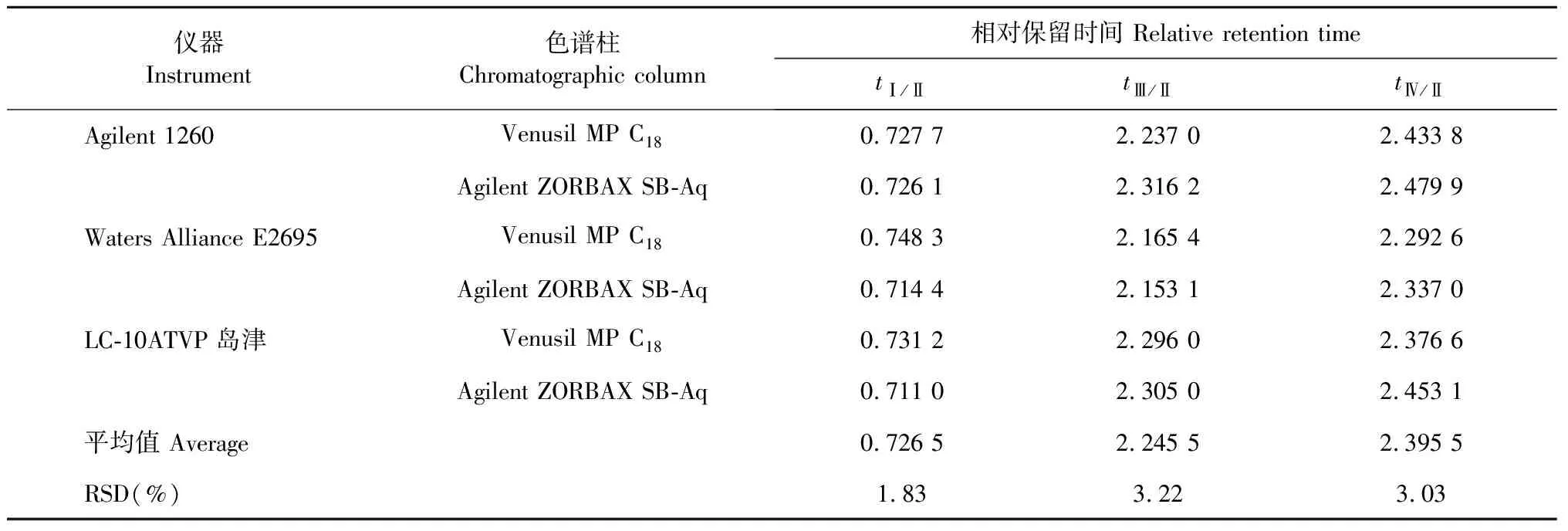
表6 不同仪器和色谱柱对相对保留时间的影响Table 6 Effects of different instruments and columns on relative retention time
2.10 样品含量测定
取11批不同产地巴戟天样品,按“2.2.2”项下方法制备巴戟天样品溶液,每个产地的药材平行制备2份样品溶液,按“2.1”项下色谱条件进样测定,记录峰面积,分别用外标法(ESM)和一测多评法(QAMS)计算各成分的含量,并将两种方法的含量测定结果进行比较,结果显示相对标准偏差(RSD)均小于2.00%,表明外标法与一测多评法测定结果之间无明显差异,说明本实验所建立的同时测定巴戟天中4种成分的一测多评法准确性良好(见表7)。

表7 不同产地巴戟天4种环烯醚萜测定结果Table 7 Determination of four iridoids in M.officinalis in different areas
3 讨论
本实验考察了提取溶剂(70%、80%、90%甲醇)、不同提取方法(超声提取法、加热回流提取法)以及不同提取时间(20、30、40 min),综合考虑各种因素,如提取方法的易操作性、运行成本、待测成分的提取率,最终确定巴戟天供试品溶液的制备方法以80%甲醇为提取溶剂、超声提取2次、每次提取时间为30 min。
本实验分别采用甲醇-磷酸水和乙腈-磷酸水作为流动相,发现以乙腈-磷酸水作为流动相时,各待测成分峰形及分离度较好,理论塔板数也较高,最终确定了流动相:乙腈(A)~0.2% 磷酸溶液 + 0.01 mol/L磷酸氢二钾缓冲盐(B),梯度洗脱(0~12 min,1% A;12~30 min,1% A→17% A;30~40 min,17% A)。
因去乙酰基车叶草苷酸对照品价格便宜、容易获得、性质稳定,故本实验将去乙酰基车叶草苷酸为内参物进行一测多评分析。
本实验建立的QAMS法,首次实现对巴戟天中4种环烯醚萜类成分水晶兰苷、去乙酰基车叶草苷酸、车叶草苷酸、车叶草苷的同时测定。通过方法学考察、相对校正因子耐用性实验以及QAMS法计算值与ESM法实测值RSD值的比较,表明本文所建立的方法操作便捷、结果准确,重复性好。解决了传统多指标成分评价模式对照品使用量大,检测成本高,部分对照品不易获得等不足。可为全面评价巴戟天的质量提供参考依据。
不同产地巴戟天各成分含量存在差异,其中以水晶兰苷的含量差异较为明显,其中第9批和第10批的水晶兰苷含量较低,其原因可能与巴戟天来源于不同产地有关。
综上所述,本实验建立的QAMS法,方法准确、可靠,重复性好,可为巴戟天的质量控制提供新的方法。
