对甲基苯磺酸在Ti/PbO2电极上的电氧化反应信息
2021-06-03叶志平周丹飞刘梓锋周青青王家德
叶志平,周丹飞,刘梓锋,周青青,王家德
(浙江工业大学环境学院,浙江杭州310032)
引 言
对甲基苯磺酸(p-TSA)分子式C7H8O3S,无水分子量172,广泛用作中间体、催化剂以及固化剂[1-2]。p-TSA易溶于水(25℃时溶解度67 g/100 ml),急性毒性(LD50)400 mg/kg(小鼠经口),进入水体会对环境造成危害,可生化性差[3-4]。
目前,高级氧化技术被广泛用于处理这类难降解有机废水[5-7]。其中,电氧化技术因无二次污染、操作简单等优点而备受研究者关注。余丽娜等[8]以Ti/SnO2-Sb2O3电极为阳极、碳纤维毡(PCF)为阴极,和活性炭填料构成三维电极,处理含p-TSA的丙烯酸丁酯生产废水,考察了废水初始COD浓度、曝气强度、极板间距对废水p-TSA等处理效果的影响。Dai等[7]以La/Gd掺杂改性Ti/PbO2电极为工作电极、Ti板为对电极,考察了初始pH、污染物初始浓度、电流密度对p-TSA降解的影响规律,在初始pH为2、污染物初始浓度500 mg/L和电流密度30 mA/cm2条件下,降解3 h后,废水中p-TSA和TOC降解率分别为94.46%和36.43%,并采用离子色谱(IC)、气相色谱与质谱联用仪(GC-MS)研究了其降解过程的中间产物。
电化学原位红外光谱技术通过红外光谱实时探测电极反应界面物质变化信息,以揭示电化学反应机理,较传统的离线色谱分析方法更为准确、全面[9-11],已广泛用于金属电极芳香族化合物的电化学行为研究。如Taleb等[9]研究了酸性体系中邻甲酚在铂电极上的开环氧化行为,以及在电极表面形成非导电性聚合物的信息。形稳电极(DSA)射信号稳定性差、电极表面处理要求高,相关原位红外光谱探测研究很少,仅Borrás等[12]结合UV-vis探讨了对氯苯酚和对硝基苯酚在Bi掺杂PbO2电极(Bi-PbO2)上的阳极反应动力学。
形稳电极Ti/PbO2由于具有析氧电位高、成本低廉、稳定性好等优点,在染料类[13]、酚类[14]、药物类[15]等工业废水处理领域得到研究和应用。为准确全面掌握这类DSA电极表面污染物反应信息,本文采用原位红外光谱法,同时结合循环伏安法、GC-MS分析,即时准确捕获p-TSA在Ti/PbO2电极表面电氧化过程的信息,揭示Ti/PbO2电极氧化对甲基苯磺酸的机理,研究其反应动力学,完善Ti/PbO2电极氧化p-TSA的界面反应描述,为后续DSA电极原位电化学反应信息研究积累经验。
1 实验材料和方法
1.1 模拟废水配制
本实验目的是探究p-TSA水相电氧化过程的信息,因此,采用p-TSA模拟废水。因氯离子在电氧化过程会干扰电极本征活性测试,模拟废水电解质采用Na2SO4溶液。p-TSA(99%,百灵威),无水Na2SO4(分析纯,上海凌峰)。实验用水由超纯水系统(Milli-Q,默克化工技术(上海)有限公司)制备,碱度调节采用NaOH(分析纯,天津化工)。对甲基苯磺酸溶液浓度为500 mg/L(根据实际废水浓度范围选择),支持电解质硫酸钠为0.1 mol/L。
1.2 循环伏安实验
循环伏安测试实验在电化学工作站(CHI660E,上海辰华)进行,三电极电解池以Ti/PbO2为工作电极,Ti片为对电极,甘汞电极(SCE)为参比电极,其中Ti/PbO2是由常规的电沉积方法制备得到[16]。扫描速率50 mV/s,扫描范围0~1.6 V。
1.3 电化学原位红外光谱实验
电化学原位红外光谱实验采用多步电位阶跃FTIRS法(MSFTIRS)和 时 间 分 辨FTIRS法(TRFTIRS),扫描在红外光谱仪(Nicolet 6700,Thermo)上进行,电解池为外反射式[17],Ti/PbO2圆盘电极为工作电极,Ti片为对电极,甘汞电极(SCE)为参比电极。
实验过程中,氮气吹扫红外仪以排除空气中CO2以及水分对实验结果判定带来的干扰。每张谱图有50张干涉图叠加,分辨率8 cm-1。所得谱图运用差谱公式计算,差谱公式如下:ΔR/R=[R(ES)-R(ER)]/R(ER),其中R(ER)和R(ES)分别是在参考电位ER和研究电位ES下采集的光谱。正向吸收峰表示研究电位下反应物的消耗,负向吸收峰对应于该电位下中间物或者产物的生成。
1.4 电解实验
电解实验的电解液为500 ml,实验温度25℃,pH取值3、7、11。Ti/PbO2阳极和Ti网阴极的尺寸均为10 cm×10 cm,电极间距2.5 cm。实验电流密度根据极限电流密度2.6 mA/cm2取值,恒电流工况下电解,分别为2.6、6.5、13、20 mA/cm2,按一定时间间隔取样,测定对甲基苯磺酸浓度[18]。平均电流效率(ACE)和能耗(EC)的计算公式分别为:
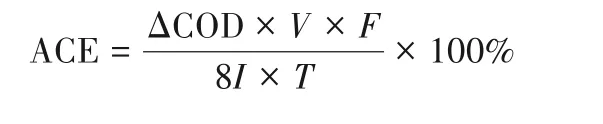
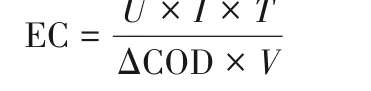
式中,ΔCOD为COD的变化,g/L;V为电解液体积,L;I为电流,A;U为槽电压,V;T是时间间隔,s或h;F是法拉第常数,96487 C/mol。
1.5 物质检测
对甲基苯磺酸采用高效液相色谱(e2695,Waters)监测,检测波长为230 nm,流动相为甲醇-三氟乙酸(0.15%)(体积比30∶70)。CODCr采用分光光度计(DR6000,HACH)监测。
中间产物采用GC-MS(7890/5975C,Agilent)检测。取50 ml电解液,用30 ml二氯甲烷分三次萃取,除去水分后得到有机相,40℃下旋转蒸发至干,加2 ml二氯甲烷溶解析出的固体,经滤膜过滤后置于样品瓶中待检测。色谱条件为:进样口温度250℃,初始柱温为40℃,保持10 min,以12℃/min的升温速率升温至100℃后保持5 min,以5℃/min的升温速率升温至200℃后保持5 min。质谱条件为:传输线温度250℃,离子源温度250℃,氦气流量1 ml/min。
2 实验结果与讨论
2.1 循环伏安实验
图1为Ti/PbO2电极在对甲基苯磺酸溶液中的循环伏安图。其中实线为Ti/PbO2电极电氧化p-TSA的循环伏安曲线,虚线为空白溶液的循环伏安曲线。与空白溶液相比,正扫过程中p-TSA在Ti/PbO2电极上出现了一个氧化峰,电位区间为0.55~0.9 V,说明Ti/PbO2电极对p-TSA具有电催化活性,直接氧化反应峰电位位于0.78 V。反扫过程中未观察到相应的还原峰,说明p-TSA在Ti/PbO2电极上发生的氧化反应是不可逆的。此外,两条曲线在1.2 V处有氧化峰,在0.9 V处有还原峰,这属于电极上PbO2/PbO的氧化还原对[19-20]。析氧电位为1.3 V。
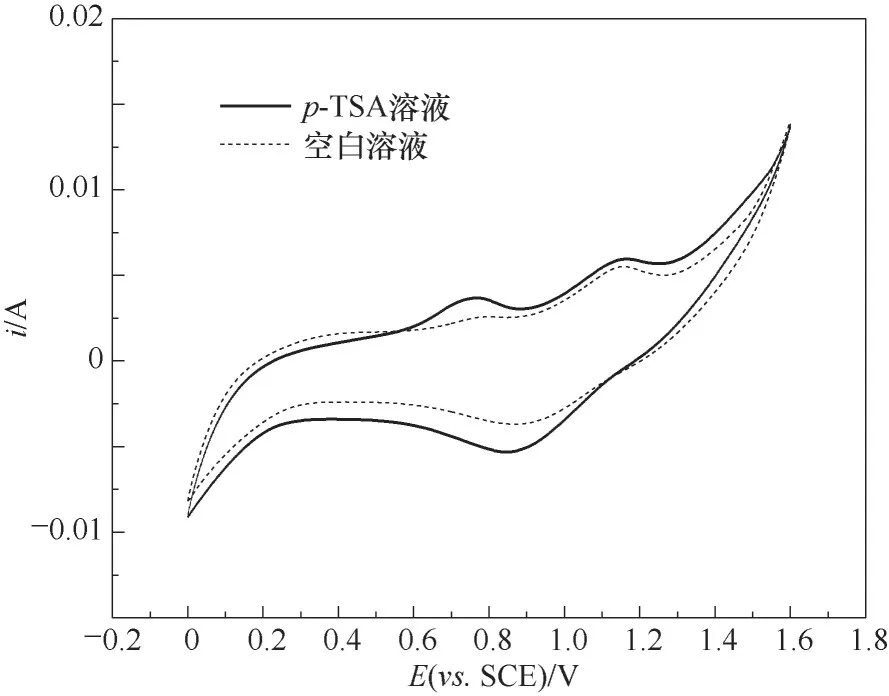
图1 Ti/PbO2电极在p-TSA溶液和空白溶液的循环伏安图Fig.1 Cyclic voltammogram for a Ti/PbO2 electrode in the presence and absence of p-TSA
2.2 电化学原位红外光谱分析
2.2.1 多步电位阶跃原位红外光谱 图2为p-TSA
在Ti/PbO2电极上的多步电位阶跃原位红外光谱图。参考电位为0 V(vs.SCE),电位从0 V阶跃到1600 mV,每个电位持续30 s。图2(b)是图2(a)电位800 mV、1300 mV谱图曲线的局部放大。
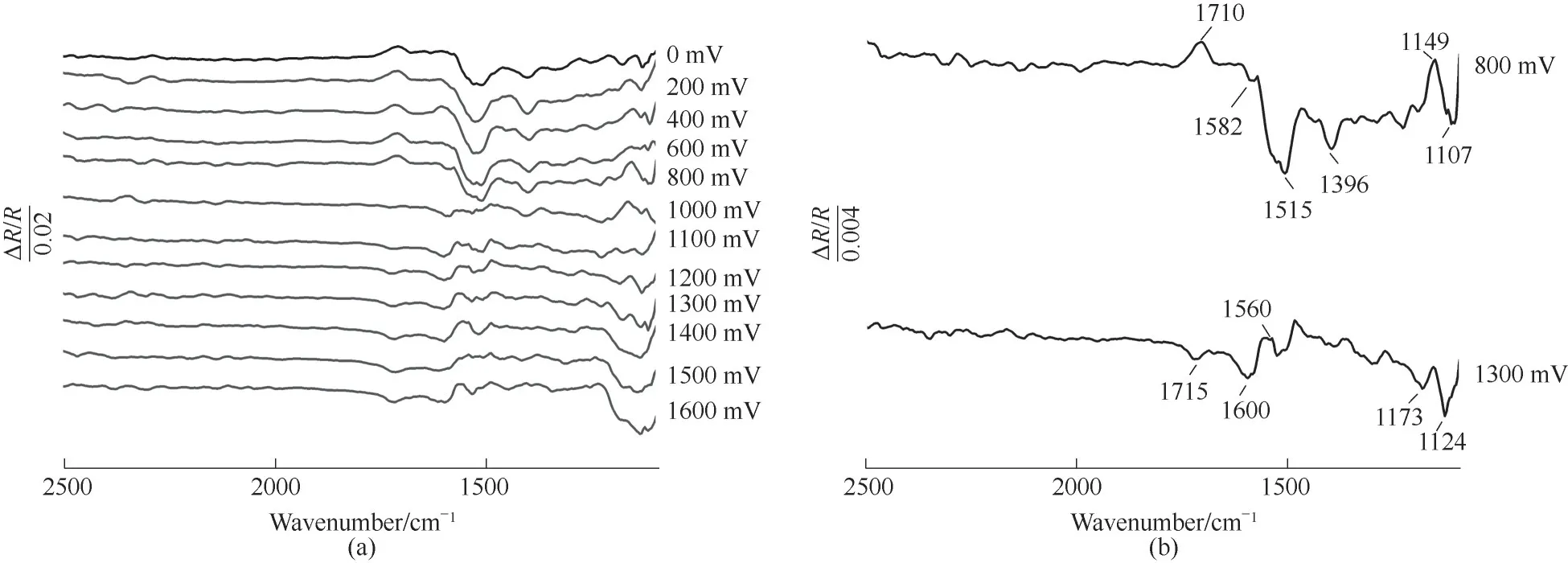
图2 p-TSA在Ti/PbO2电极上的多步电位阶跃原位红外光谱图Fig.2 MSFTIRspectra of p-TSA on Ti/PbO2 electrode
低电位(如800 mV)时,可以观察到正向吸收峰1710、1149 cm-1和 负 向 吸 收 峰1582、1515、1396、1107 cm-1。其中1710 cm-1属于结晶水变角振动[21],1149 cm-1属于p-TSA的磺酸OSO反对称伸缩[21],反映了磺酸基的脱落。张凌等[22]采用MOPAC/PM3算法发现C—S键电子云密度最高,最易受到攻击,这与实验在低电压下仅出现与物质相关的磺酸基的消耗一致。负向峰1582、1515、1396 cm-1均属于苯环骨架振动峰[23-25],1107 cm-1属于芳环CH面内弯曲。随着电位增大,负向吸收峰强度先增大后减小,说明p-TSA生成了不带磺酸基的苯环类物质,并且该物质在薄层溶液中发生了先积累后消耗的变化。
当电极电位正移到1000 mV以上(如1300 mV)时,吸收峰强度随着电位增大而增强变大,p-TSA苯环结构发生了开环(正向峰1560 cm-1)、降解;同时观察到负向吸收峰1715、1600、1173和1124 cm-1,分别属于羧酸C O伸缩振动、羧酸根COO反对称伸缩振动[26-28]、酮羰基和甲基C—C伸缩振动以及醇C—OH伸缩振动[29],即p-TSA开环生成了羧酸、酮和醇。
2.2.2 时间分辨原位红外光谱 图3(a)、(b)为p-TSA溶液在电位600 mV和900 mV下电解300 s得到的时间分辨原位红外光谱图。参考电压均为0 V(vs.SCE)。
由图3(a)、(b)可知,峰的强度随电解时间增加而增强。电解时间300 s,电位600 mV情况下,出现 负 向 峰3750、1555、1515、1405、1315、1207、1157、1110 cm-1,其中较为宽泛的负向峰3750 cm-1属于氢氧化物O—H伸缩,负向峰1555、1515 cm-1属于苯环的骨架振动[30],1157、1110 cm-1属于芳环C—H面内弯曲,1207 cm-1属于芳环和醛羰基C—C伸缩,1405、1315 cm-1可能分别属于羧酸的COH面内弯曲振动[26]和C—OH伸缩振动。由于尚未到达开环电压,因此初步判断醛和羧酸均为苯环上的取代基。电位900 mV时,即发生了开环(相较于1000 mV),这是由于电解时间更充分的缘故,新出现了属于羧酸C—OH伸缩振动的负向峰1273 cm-1,即产生羧酸产物。
2.3 反应机理
在电流密度13 mA/cm2下电解3 h,取电解液50 ml,经预处理后检测产物。p-TSA电氧化过程的中间产物结果见图4。如图4显示,p-TSA电氧化过程涉及的大分子物质有苯甲醇、苯甲醛、苯甲酸、对甲酚,小分子物质有乙酸、丙酮、1-羟基丁酸等。
结合上述红外谱图、GC-MS分析,以及文献报道[8],得出了p-TSA在Ti/PbO2电极上的可能降解路径见图5。降解路径有两条:一条为羟基取代磺酸基生成对甲酚,继续氧化生成对苯二酚,然后发生开环,但是红外薄层溶液实验未检测到该路径,可能与中间产物浓度低等原因[31]有关;另一条为磺酸基脱落后,羟基自由基攻击苯环的甲基侧链,随后发生开环,逐渐生成苯甲醇、苯甲醛、苯甲酸,随后在高电压处发生开环,形成脂肪酸、酮、醇。
2.4 反应动力学
如图6所示,p-TSA在Ti/PbO2为阳极、Ti网为阴极的电解槽体系中,反应速率随电流密度增大而快速上升,表观速率常数K与电流密度j呈线性关系:
K=0.0062j-0.0071,R2=0.98 (1)
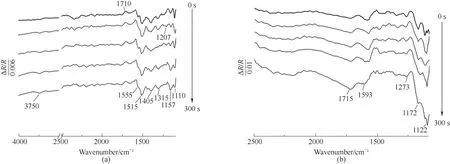
图3 600 mV(a)和900 mV(b)时p-TSA的时间分辨原位红外光谱图Fig.3 TRFTIRspectra of 600 mV(a)and 900 mV(b)of p-TSA

图4 p-TSA电氧化的主要中间产物Fig.4 Main intermediates formed in the p-TSA degradation by electro-oxidation
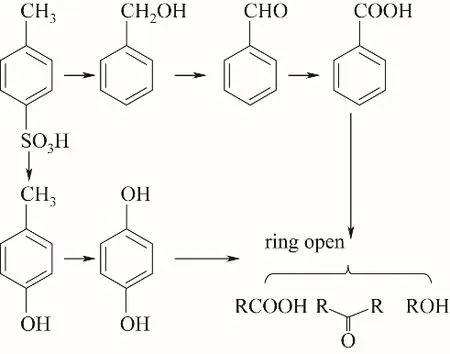
图5 p-TSA在Ti/PbO2电极上降解路径Fig.5 The pathway of p-TSA degradation on Ti/PbO2 electrode
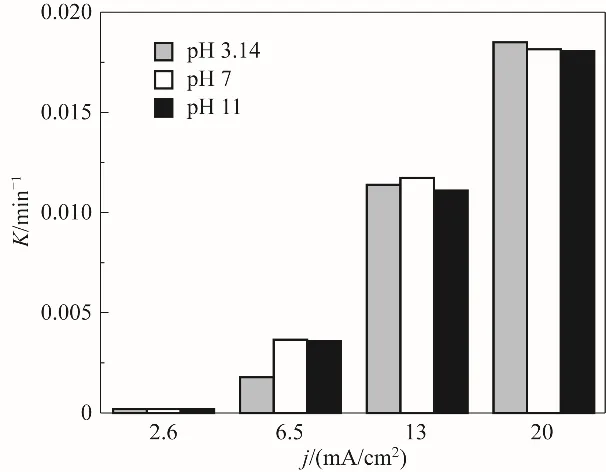
图6 电流密度、pH对表观速率常数(K)的影响Fig.6 The influence of current indensity and pH on apparent rate constant(K)
相较电流密度,pH对表观速率常数的影响不大。随着电流密度增大,最适pH由中性转为酸性,分析认为,p-TSA溶液中存在形态受pH影响,酸性溶液主要以分子形式存在,碱性溶液以阴离子形式存在;而且,酸性条件抑制析氧,碱性条件促进析氧。因此,电流密度6.5、13 mA/cm2时,最适pH为中性;电流密度20 mA/cm2时,最适pH转为酸性。
比电荷是处理单位水量输入的电量(单位A·h/L),用于描述电氧化去除废水中污染物涉及的能量。一般地,污染物去除量随比电荷增加而增加,同时,平均电流效率随比电荷增大而减小,能耗也呈增大趋势。如图7所示,当比电荷从1.3 A·h/L逐渐增大到7.8 A·h/L时,化学需氧量(COD)减少到原先的45%,平均电流效率从22.19%降至19.13%,是在流通系统下钌钛电极氧化芳香族磺酸类化合物的4倍[32](5.1%)。比电荷5.2 A·h/L起,能耗开始急剧上升,原因是随电解时间延长,降解过程中电极极化以及析氧副反应加剧。
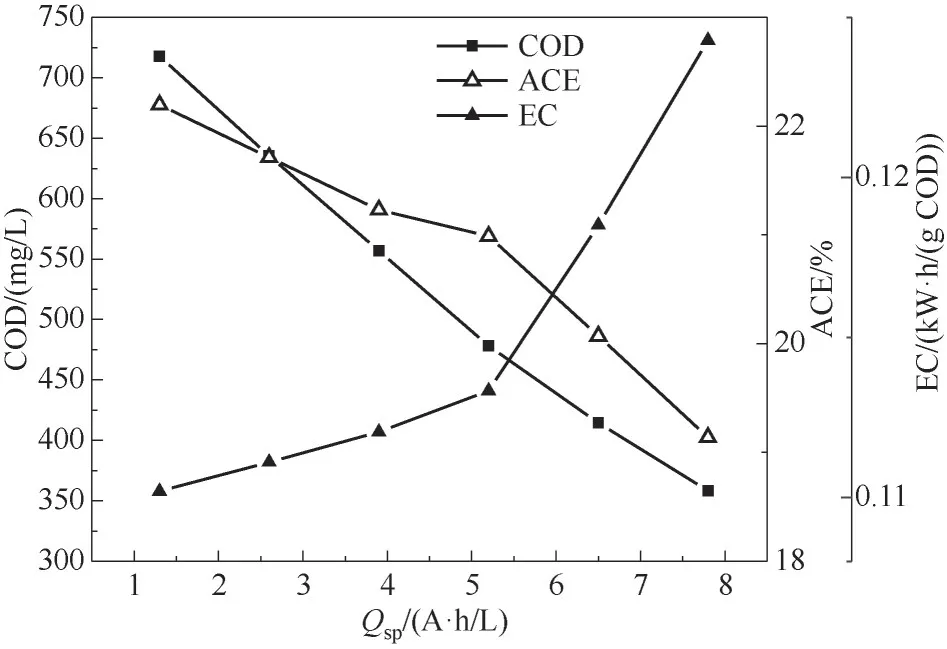
图7 比电荷(Q sp)对p-TSA去除、能耗(EC)和电流效率(ACE)的影响Fig.7 The influence of specific electric charge(Q sp)on p-TSA romoval,energy consumption,and average current efficiency
3结 论
本文研究了p-TSA在Ti/PbO2电极上的原位红外光谱信息。Ti/PbO2对p-TSA具有催化活性,直接氧化电位区间为0.55~0.9 V。多步电位阶跃FTIRS法和时间分辨FTIRS法分析显示,低电位(<1000 mV)处,p-TSA主要发生了磺酸基的脱落和苯环侧链甲基的氧化;高电位(>1000 mV)下,p-TSA发生苯环破坏,生成了酸、醇、酮。
电流密度和pH影响电氧化速率,相对而言,电流密度的影响更显著,表观速率常数K与电流密度j呈线性关系。污染物去除量随比电荷增加而增加,比电荷从1.3 A·h/L逐渐增大到7.8 A·h/L,化学需氧量(COD)减少到原先的45%,同时能耗呈增大、平均电流效率呈下降趋势。
