基于生物信息学筛选甲状腺癌关键基因和通路
2021-06-01
(复旦大学附属闵行医院病理科,上海市 201199)
甲状腺癌(thyroid carcinoma,TC)是内分泌系统最常见的恶性肿瘤,也是近20年来发病率增长最快的实体恶性肿瘤[1]。然而,Davies等[2]提出TC发病率的上升可能有两个原因:一个是过度诊断,另一个是甲状腺乳头状微小癌发病率增加。目前,超声引导下细针穿刺活检是评价甲状腺结节的金标准,另外还尝试了ECT、CT等新技术[3]。
近年来,随着基因芯片和高通量测序技术的发展,已经筛选出一些在癌症发生和发展过程中具有重要意义的基因。许多研究分析了GEO(https://www.ncbi.nlm.nih.gov/geo)或/和TCGA(https://cancergenome.nih.gov/)的数据,以寻找对甲状腺癌的诊断和预后有前景的生物标志物。例如,Min等[4]分析了TCGA的数据,确定了一些预后标志物,如SOX4、RARA,这些标志物可作为甲状腺乳头状癌(thyroid papillary carcinoma,PTC)的治疗靶点。Wu等[5]基于TCGA数据库进行富集分析发现,AP-2α mRNA的表达可能是PTC患者生存的一个独立预后指标。由于独立研究样本的异质性,目前的研究结果往往不一致。本文采用生物信息学工具对GEO数据库中甲状腺癌表达谱芯片进行数据分析,为今后提高甲状腺病变的诊断水平和基因靶向治疗提供新的视角。
1 材料和方法
1.1 材料
基因表达谱芯片数据来源于GEO数据库中甲状腺癌数据集,编号分别为GSE33630、GSE65144和GSE29265,包括101例甲状腺癌组织和78例正常组织。数据集均基于GLP570芯片平台(HG-U133_Plus_2;Affymetrix Human Genome U133 Plus 2.0 Array)。
1.2 数据处理及筛选差异表达基因
3组原始数据集下载后利用R软件进行合并、标准化及表达值计算等处理,数据采用Fold-change和T-test进行差异性基因筛选,筛选条件定义为|logFc|≥1、P<0.05,筛选3组数据集中共有的差异基因作为最终差异表达基因进行后续分析。
1.3 差异表达基因的功能富集分析
基因本体论(GO)是一种生物学模型框架,由三大板块构成:分子功能(molecular function,MF)、细胞成分(cellular component,CC)和生物学过程(biological process,BP)。京东基因组百科全书(KEGG)是从分子水平系统理解生物系统和基因组功能的信息资源。使用R包clusterProfiler进行差异表达基因(differentially expressed genes,DEGs)的GO分析,KOBAS3.0进行KEGG通路分析。
1.4 差异表达基因编码蛋白的相互作用网络分析
STRING数据库(https://string-db.org/)是一个用于预测EMBL中基因互作网络的分析数据库,该数据库聚集来自蛋白的相互作用网络(protein protein interaction network,PPI)数据库的提取结果。采用Cytoscape(www.cytoscape.Org)中MCODE插件对网络模型进行评价,选择排名前3的模块中的基因进行通路富集分析。
1.5 关键基因的筛选
将PPI得到的结果导入Cytoscape 3.8.2软件中的cytahubba程序包进行分析,将cytohubba中12种算法各自获得的前30个关键基因求交集,选定在12种算法中出现8次及以上的基因为关键基因。
1.6 关键基因的验证
利用TCGA_TC数据集的505个TC样本和59个正常样本验证关键基因的表达,然后采用Kaplan-Meier(Km)曲线评估关键基因表达水平与TC患者生存时间的关系。
1.7 统计学方法
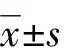
2 结 果
2.1 甲状腺癌DEGs筛选
经R软件分析,分别从GSE33630、GSE65144、GSE29265中得到1 145、2 552、792个DEGs,取3个数据集DEGs进行分析,得到相同DEGs共有410个(图1),其中TC样本中表达量上调的基因有159个,下调的基因有251个(图2)。
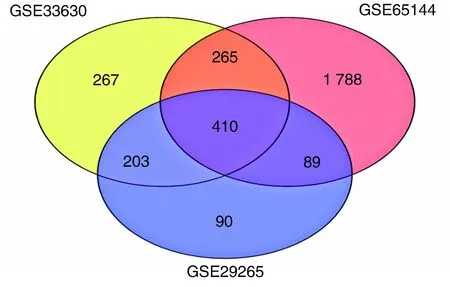
图1 3组基因芯片数据的DEGs关系图
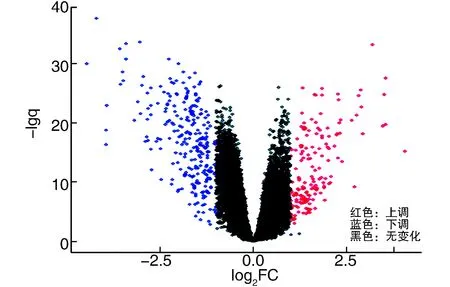
图2 DEGs火山图
2.2 DEGs的GO富集分析结果
上调基因主要富集在内胚层细胞分化、胶原原纤维组织、透明质酸结合等生物学过程(图3A),下调基因主要富集在甲状腺激素生成、甲状腺激素代谢过程、过氧化物酶激活等生物学过程(图3B)。
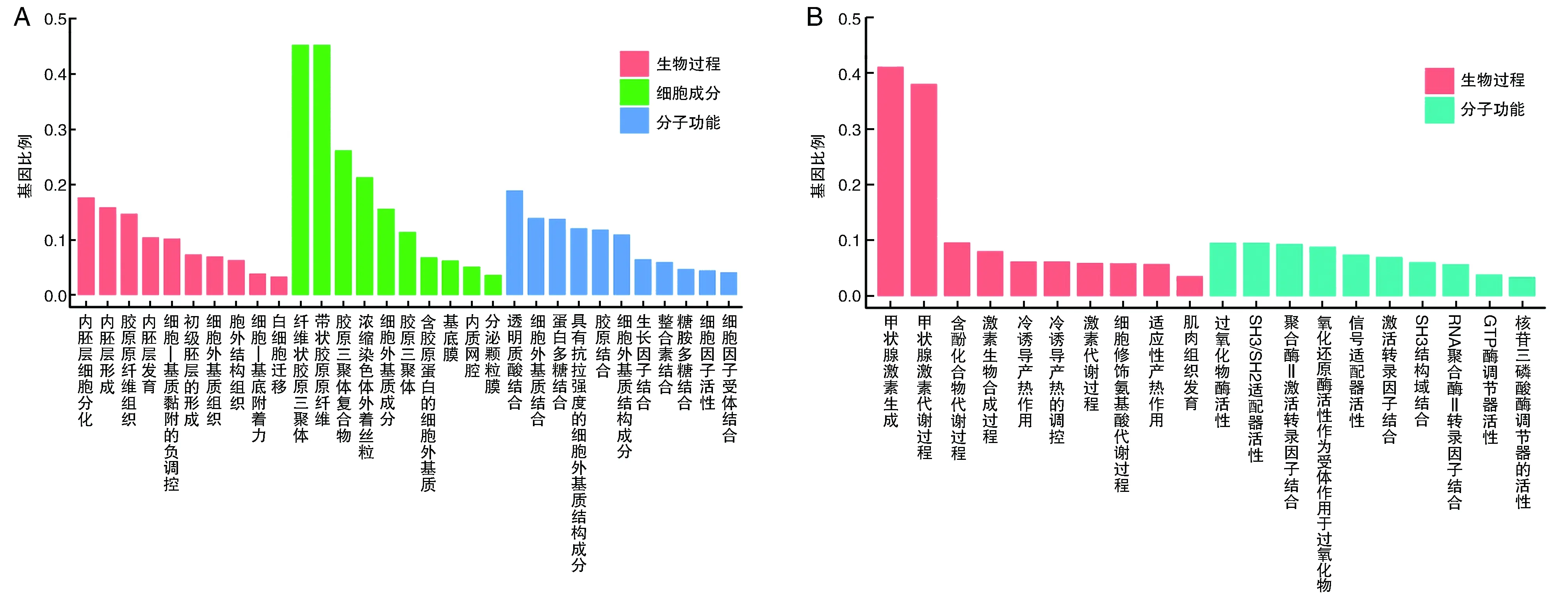
图3 DEGs的GO富集分析A为上调基因GO富集分析结果;B为下调基因GO富集分析结果。
2.3 DEGs的PPI分析结果
使用插件MCODE对字符串网络进行分析,筛选出最重要的3个模块(图4)。模块A由22个节点和291条边组成;模块B由17个节点和67条边组成;模块C由8个节点和26条边组成。
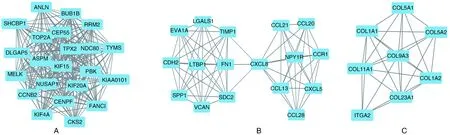
图4 PPI分析图A为细胞周期、p53信号通路;B为病毒蛋白与细胞因子-细胞因子受体相互作用;C为蛋白质的消化和吸收关系
2.4 关键基因筛选及KEGG通路分析
共获得14个关键基因,分布在30条KEGG信号通路中,与所有DEGs富集的KEGG信号通路排名前20位共同的信号通路有6条(表1,下划线基因为关键基因),其中上调的基因有12个,分别是CCNB2、FN1、MMP9、TIMP1、CXCL8、VCAN、EVA1A、LGALS1、KIF15、KIF20A、KIF4A、TOP2A,下调的基因有2个,分别是JUN、SDC2。其中TOP2A、KIF15、CCNB2、KIF4A位于模块A,FN1、LGALS1、TIMP1、VCAN、SDC2位于模块B。

表1 关键基因富集的KEGG信号通路
2.5 关键基因的验证和预后标志物的预测
分析来自TCGA_TC数据集的505个TC样本和59个正常样本14个关键基因的表达水平,其表达均与DEGs分析结果一致,其中MMP9、SDC2、KIF15和VCAN4个基因影响患者生存率(MMP9P=0.11、VCANP=0.11、KIF15P=0.042、SDC2P=0.061)(图5)。从生存曲线结果可以看出,随着基因的表达增高患者的生存率明显下降,即这些基因的高表达促进了甲状腺癌的发展,而且从生存曲线可以看出,4个基因低表达时的生存曲线与高表达时相贴近,生存率均较高,但长期随访,则在低表达时患者生存率较高,即这些基因对甲状腺癌的发展影响较大,对患者生存时间影响较大,提示可以作为TC的潜在预后标志物。
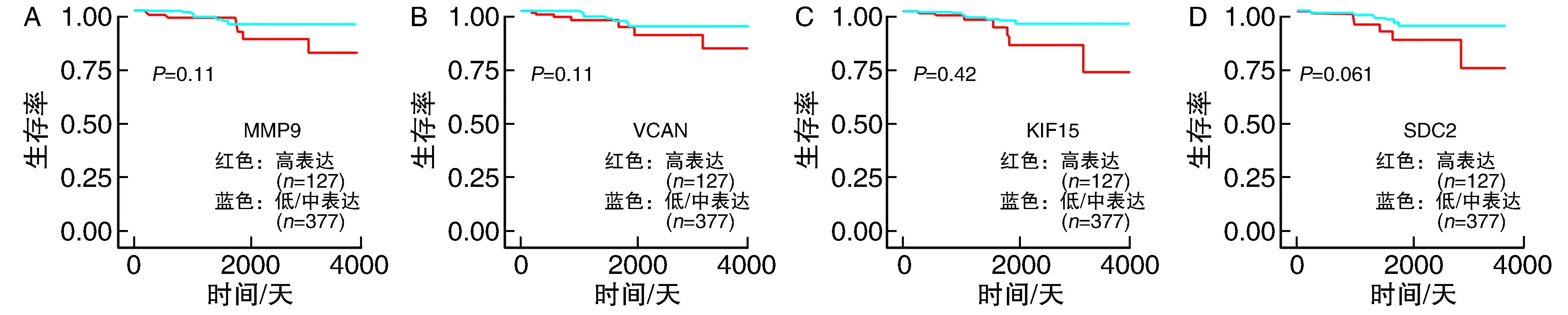
图5 TCGA数据库中MMP9、VCAN、KIF15、SDC2对甲状腺癌患者生存的影响
3 讨 论
目前,有关TC组织与正常组织基因表达差异的研究不多。寻找肿瘤与正常组织之间的DEGs有助于进一步了解TC的发病机制,为TC的术前诊断提供生物标志物和治疗靶点。本研究使用GEO数据库中同一平台的数据,以人TC组织和正常组织为研究对象,用生物信息学方法进行了深入分析。
从TC的3个基因芯片数据集中鉴定出410个DEGs,其中包括159个上调基因和251个下调基因。GO分析表明DEGs可能对内胚层细胞分化、胶原纤维组织、甲状腺激素生成、甲状腺激素代谢过程等生物学过程都有一定影响。KEGG通路分析中,DEGs主要富集于癌症中的蛋白聚糖、P53信号通路、ECM-受体相互作用、细胞周期等信号通路。肿瘤细胞的生长由各种生长因子、激素及细胞外基质等物质构成的微环境维持,这些因素的改变会使肿瘤的发生、发展过程及对药物敏感性发生变化。因此,监测以上信号通路可能有助于预测甲状腺癌的进展及其对药物的敏感性。
本文筛选出14个关键基因CCNB2、FN1、MMP9、TIMP1、CXCL8、VCAN、EVA1A、LGALS1、KIF15、KIF20A、KIF4A、TOP2A、JUN、SDC2,发现TOP2A、KIF15、CCNB2、KIF4A、FN1、LGALS1、TIMP1、VCAN、SDC2位于PPI分析得出的3个显著模块中,采用TCGA_TC数据集的505个TC样本和59个正常样本进行验证,这14个关键基因的表达水平均与DEGs分析结果一致:JUN和SDC2在TC中下调,其余12个关键基因上调。其中MMP9、SDC2、KIF15和VCAN影响患者生存率,可以作为TC的潜在预后标志物。这些基因可能参与TC的各个阶段,并在3个数据集中共表达。
已有研究表明,关键基因在多种癌症相关的生物学过程中发挥着重要作用。MMP9可降解细胞外基质中的明胶、多种胶原及弹性纤维,尤其是Ⅳ型胶原蛋白,在癌细胞的浸润和转移中发挥着重要作用。Kalhori等[6]研究表明MMP9参与了S1P诱导的滤泡型甲状腺癌细胞的侵袭。VCAN位于染色体5q12-14,它的异常表达与多种肿瘤不良预后密切相关,体内外研究表明,VCAN调节多种细胞过程,包括肿瘤表型和性质、耐药性的发展和肿瘤基质血管生成等[7]。Zhao等[8]发现PTC中VCAN出现反复发生的突变。还有研究发现MIR-135a-5p通过靶向VCAN抑制甲状腺癌细胞的增殖、侵袭和迁移[9]。另外,CCNB2属于B类细胞周期蛋白家族,Wang等[10]提出miR-205通过靶向CCNB2抑制甲状腺癌细胞的增殖和迁移。FN1是细胞外基质的基本成分,研究发现FN1在甲状腺癌中过表达[11],也有建议将FN1作为识别不明FNAB患者恶性病变的分子标志物[12]。Sponziello等[13]研究表明,FN1的过度表达使PTC更具侵袭性,并可能促进甲状腺肿瘤的进展。TIMP1作为金属蛋白酶(MMPs)的抑制剂被熟知,TIMP1与MMPs之间的动态平衡,维持着细胞外基质的稳定,与肿瘤转移密切相关。Maeta等[14]研究发现,TIMP1在PTC中表达上调,且与肿瘤大小、淋巴结转移和临床分期显著相关。Hawthorn等[15]提示TIMP1的高表达可作为PTC诊断的候选分子标志物。CXCL8是一种由正常细胞和甲状腺癌细胞分泌的趋化因子,有研究证明了较高的肿瘤内CXCL8水平与甲状腺癌更具侵袭性的病程之间存在正相关关系[16]。EVA1A是一种参与细胞程序性死亡的新基因,EVA1A的表达是PTC肿瘤、淋巴结转移的独立危险因素,EVA1A的下调可以抑制PTC细胞的集落形成、增殖、迁移和侵袭[17]。LGALS1是半乳糖结合蛋白家族的15个成员之一,编码为Galectin-1。Galectin-1与甲状腺癌的发展密切相关,已被认为是鉴别良恶性结节的可靠生物标志物[18]。
同时KIF15、KIF20A、KIF4A、TOP2A、JUN和SDC2在TC中的作用尚不清楚。一些文献已经证明了这些基因在其他癌症中的功能,TOP2A编码DNA拓扑异构酶,对DNA转录和复制至关重要[19]。SDC2能够由肿瘤细胞合成,并通过相关信号转导通路和肿瘤源性成纤维细胞的活化,在肿瘤源性血管生成和肿瘤细胞侵袭转移中起到了重要的调控作用[20]。细胞性Jun(c-Jun)和病毒性Jun(v-Jun)均可诱导肿瘤转化[21]。KIF15、KIF20A、KIF4A为驱动蛋白超家族,是一类参与细胞内组分运输的动力蛋白,负责将细胞内组分沿微管组成的细胞骨架运输至特定的位置[22]。推测这些基因通过细胞周期调控、调节细胞黏附和其他机制来调控肿瘤的发生和发展。它们可能是甲状腺癌潜在的新的治疗靶点及生物标志物。
综上所述,本文通过生物信息学方法分析甲状腺癌基因表达谱芯片数据,筛选了14个关键基因和通路,其中4个基因可作为潜在预后标志物,可能有助于甲状腺癌的早期分子诊断与基因靶向治疗。然而,所有的预测结果还需要其他体外和体内实验数据的证实。
